Date: Tue, 15 Jun 2021 23:04:45 +0530
Dear Amber Experts,
I am using Amber(pmemd.cuda) in a cluster with 64 nodes, each containing 2
Nvidia Tesla K40m (not peer-to-peer connected). This is a shared cluster
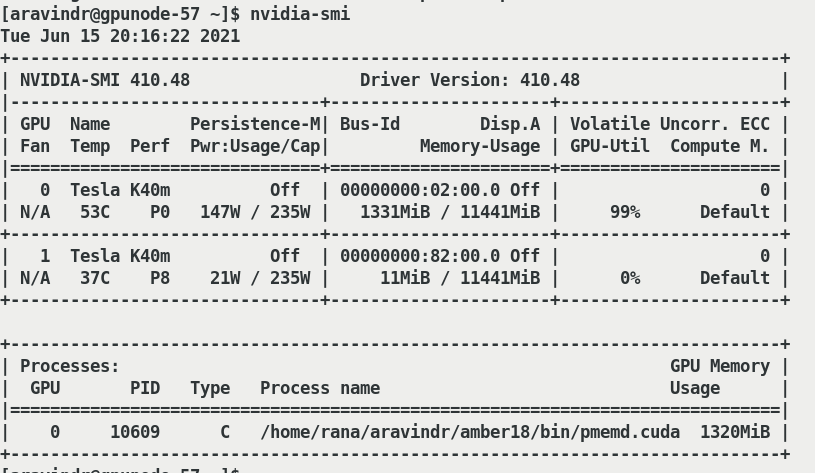
across the institute. When amber runs in a single GPU with single core
separately it utilises 99% of GPU load and runs properly(attached
Only_amber.png).
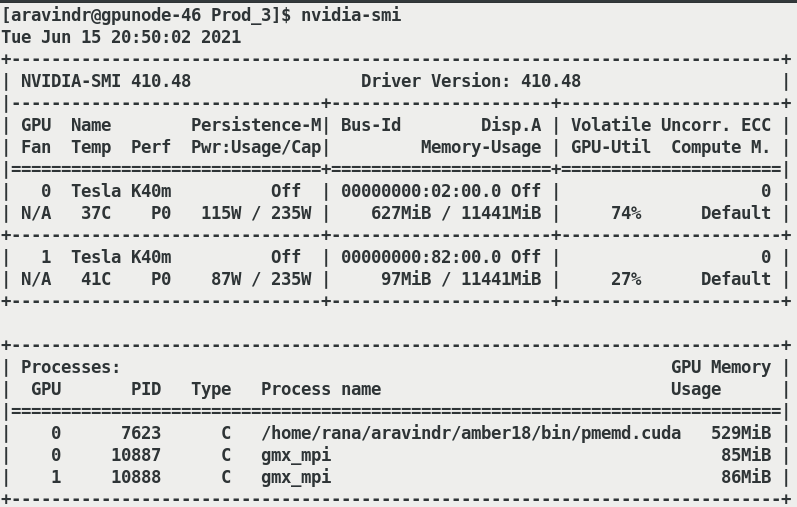
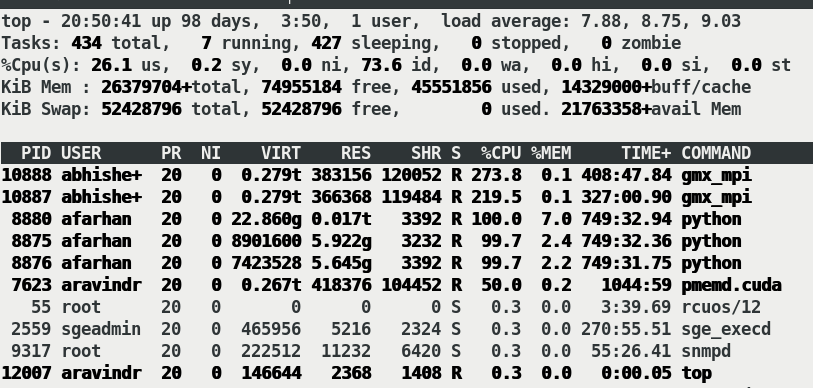
But when the gromacs job is submitted to the same node, the GPU utilisation
decreases drastically (attached Amber_&_gromacs.png) and thereby the speed
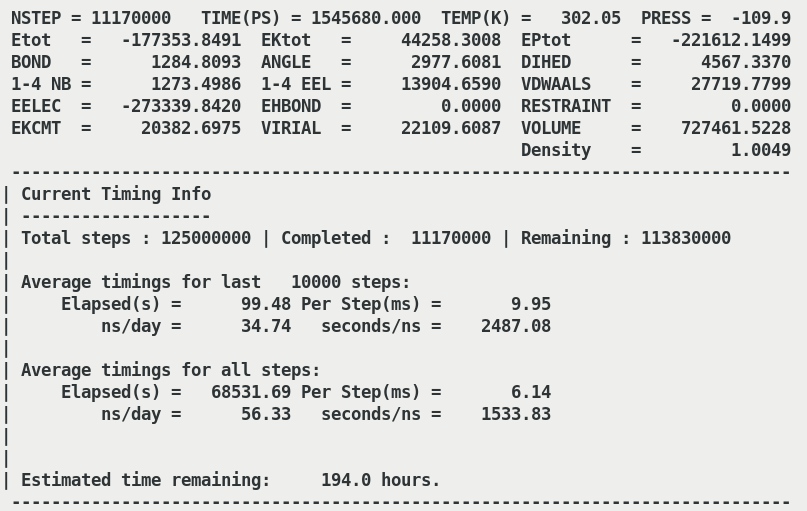
at which the job runs (attached Speed.png). Whereas gromacs jobs utilise
the GPU completely.
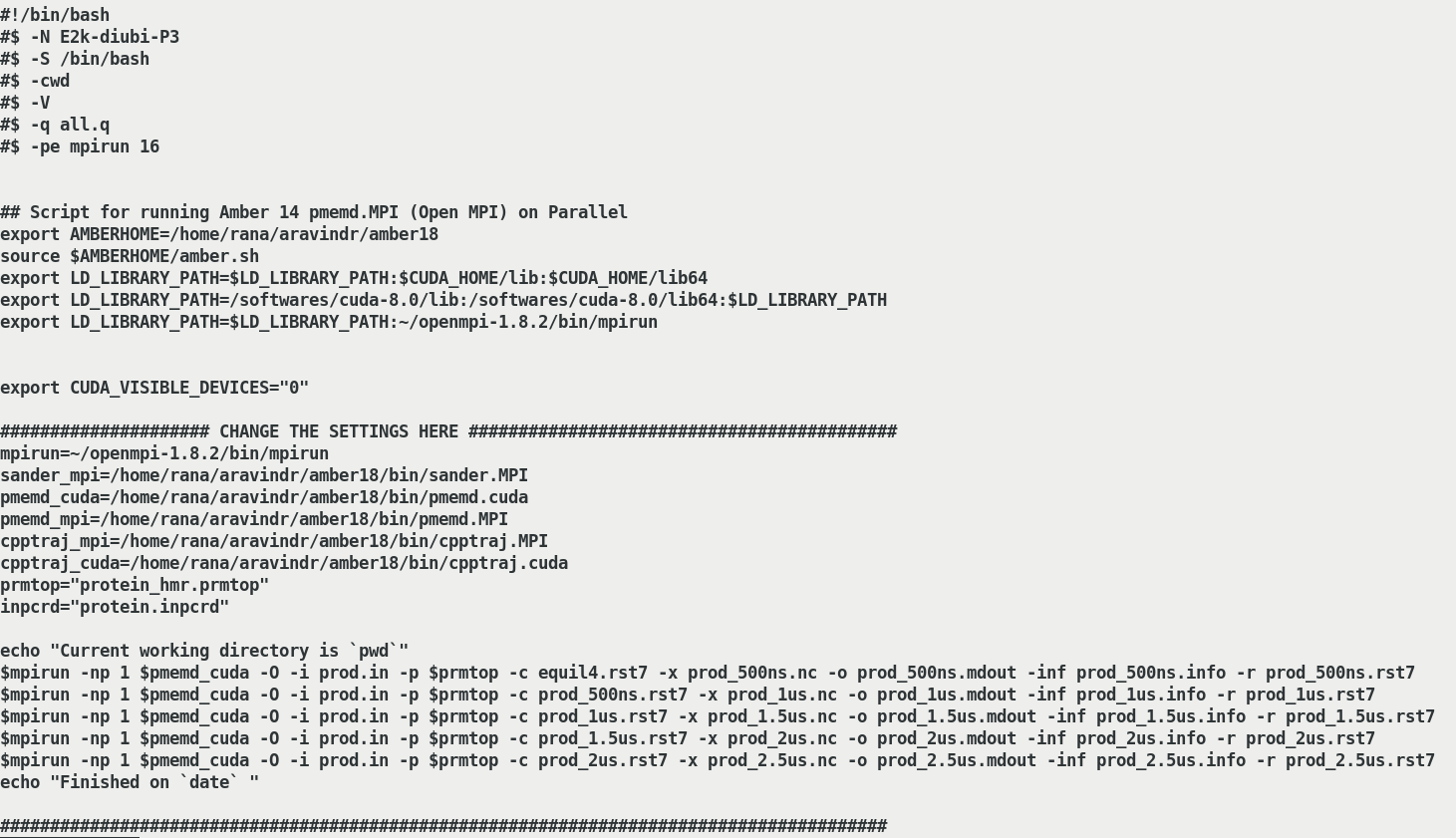
The script I use to submit the job attached herewith(Input_script.png)
along with top command results(Top_results.png).
Also, scheduler allows gromacs to run on the GPU when amber is already
running whereas not the vice-versa.
What is the reason for the decrease in GPU utilisation?
Thanks in advance,
Aravind R
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Amber___gromacs.png)
(image/png attachment: Input_script.png)
(image/png attachment: Speed.png)
(image/png attachment: Only_amber.png)
(image/png attachment: Top_results.png)
