Date: Fri, 18 Dec 2020 16:36:25 +0900
Dear All,
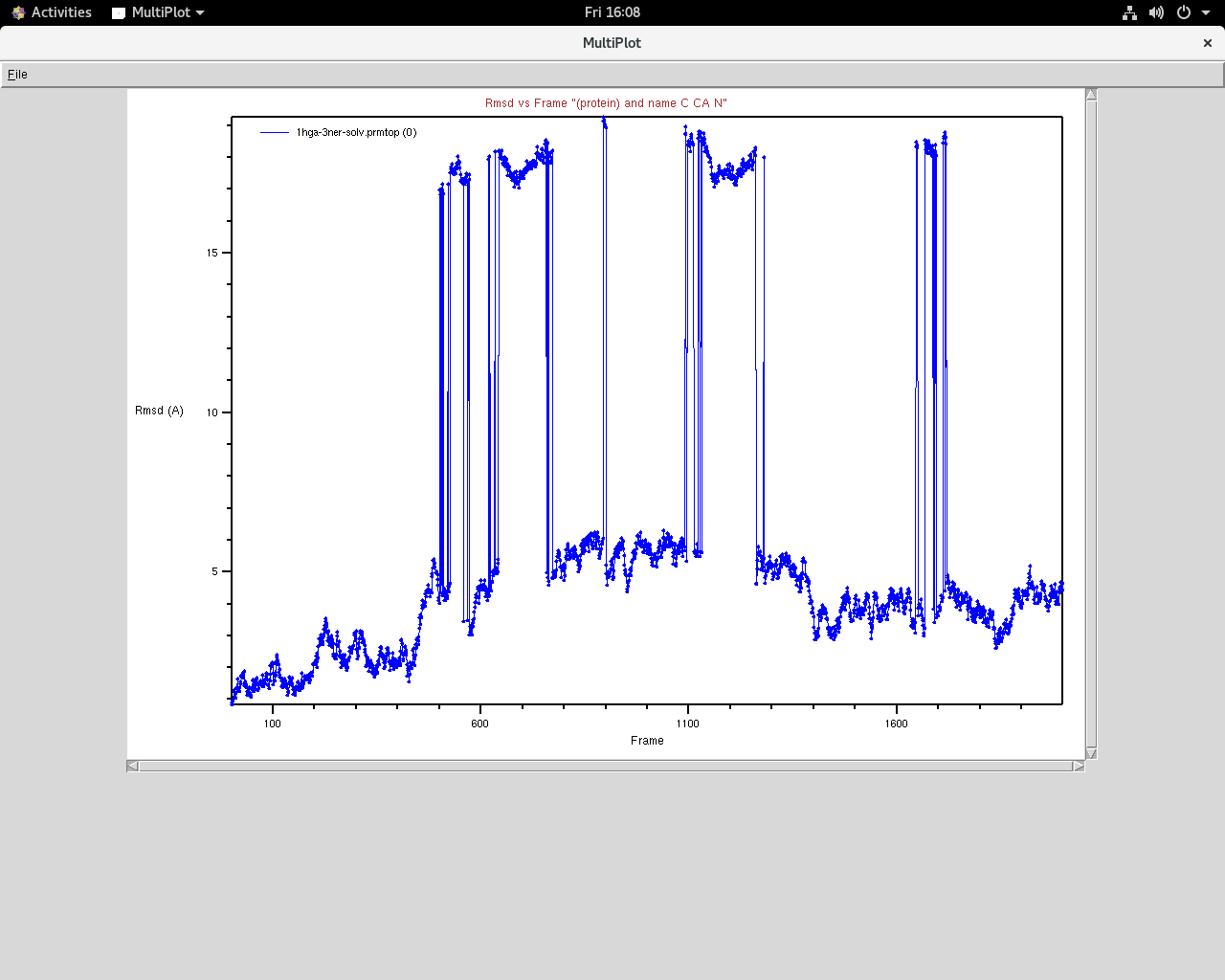
I'm simulating a complex of two metalloproteins (120 ns trajectory).
After about 80 ns, I'm observing a sudden jump in rmsd which looks
unphysical but I'm not sure if it is due to some issue with wrapping.
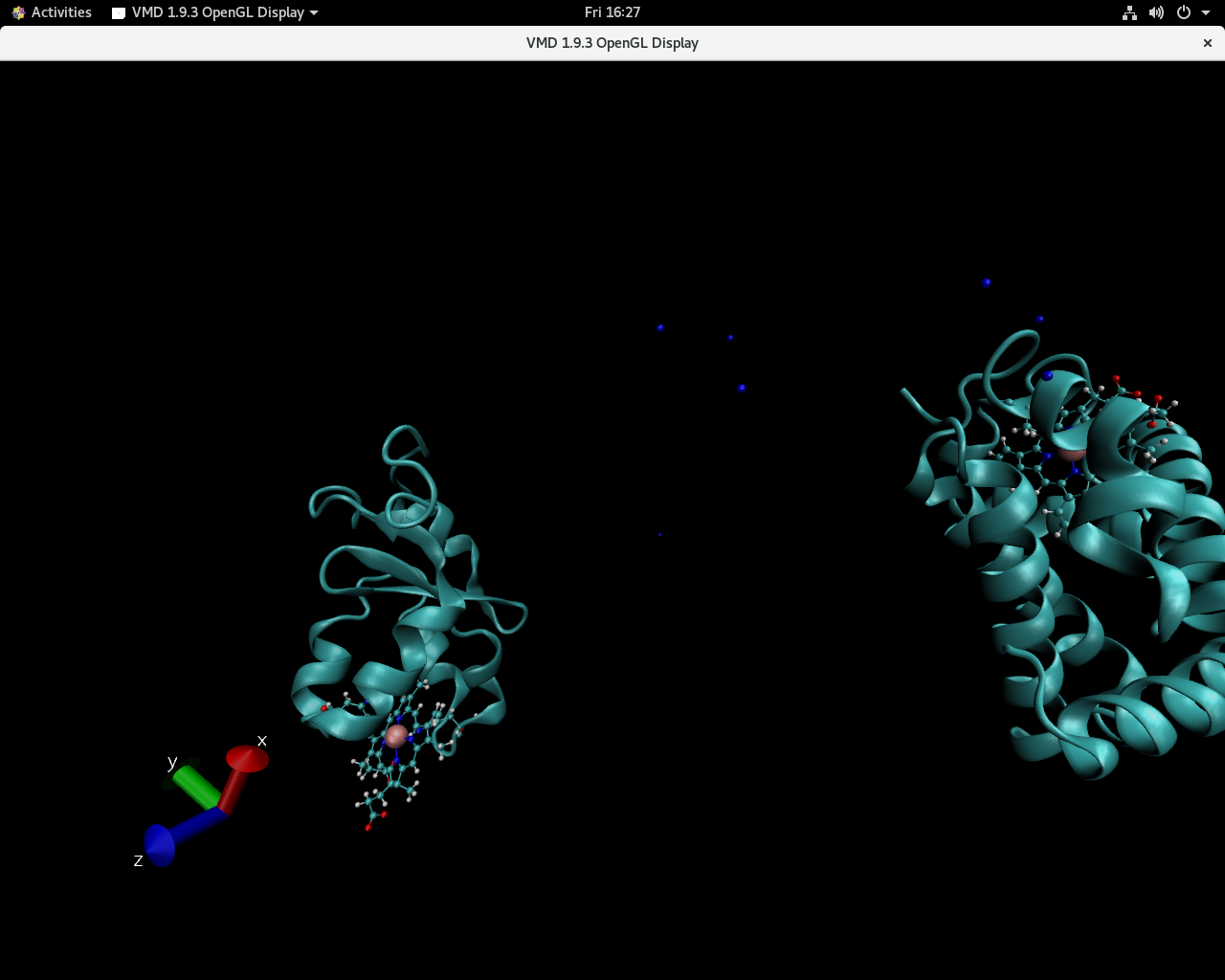
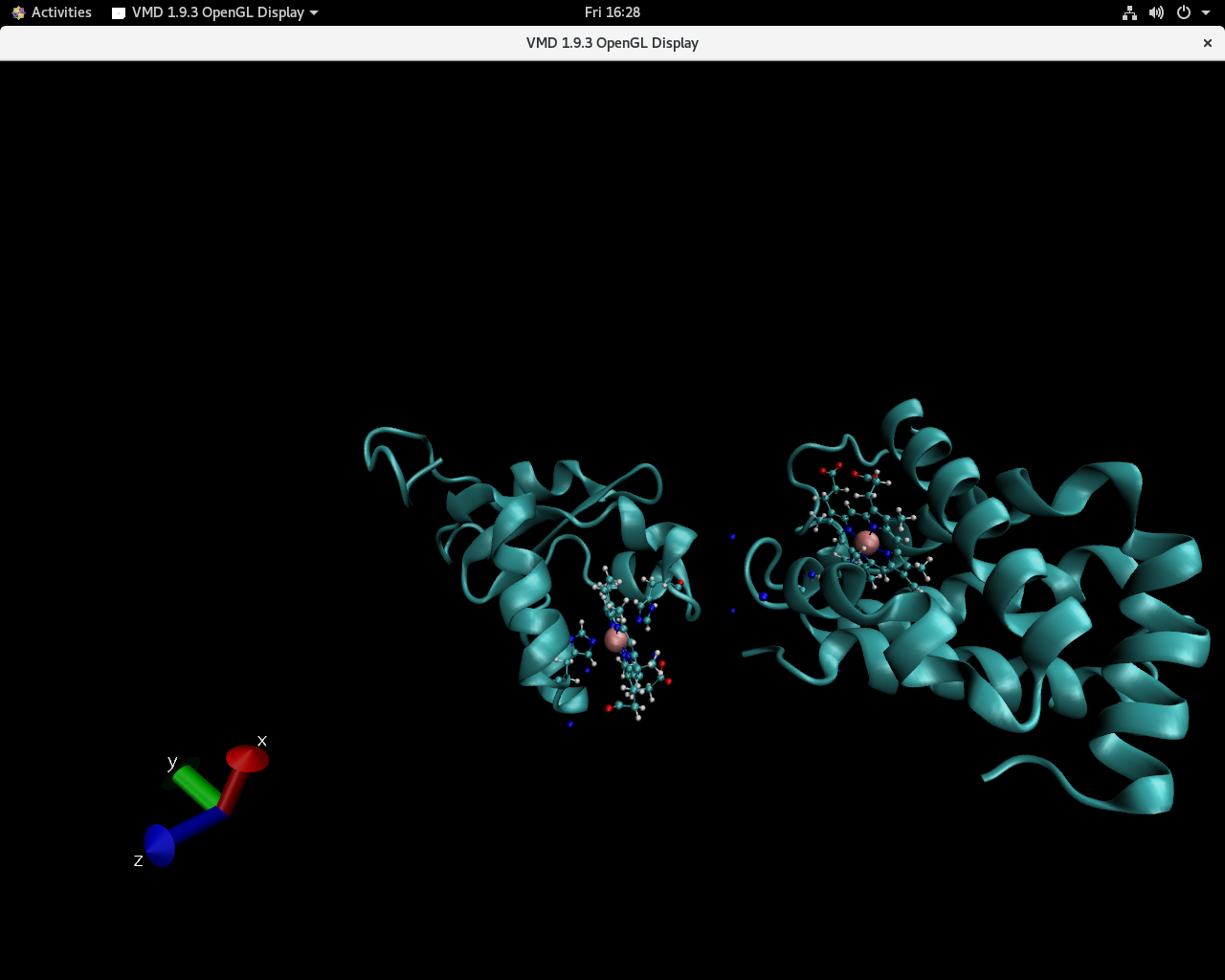
Visualization in VMD shows that one of the protein suddenly changes
position.
Both proteins seem to be well within the solvent box, thus I'm unable to
guess what is the problem here.
I have pasted two snapshots below in which the sudden jump is apparent.
Please suggest to me ways in which I can check if these jumps are genuine
or some type of artifact in the simulation.
Thank you and best regards.
[image: image.png]
[image: image.png]
[image: image.png]
-- Regards, Dr. Vaibhav A. Dixit, Visiting Scientist at the Manchester Institute of Biotechnology (MIB), The University of Manchester, 131 Princess Street, Manchester M1 7DN, UK. AND Assistant Professor, Department of Pharmacy, ▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄ Birla Institute of Technology and Sciences Pilani (BITS-Pilani), VidyaVihar Campus, street number 41, Pilani, Rajasthan 333031. India. Phone No. +91 1596 255652, Mob. No. +91-7709129400, Email: vaibhav.dixit.pilani.bits-pilani.ac.in, vaibhavadixit.gmail.com http://www.bits-pilani.ac.in/pilani/vaibhavdixit/profile https://www.linkedin.com/in/vaibhav-dixit-b1a07a39/ ORCID ID: https://orcid.org/0000-0003-4015-2941 http://scholar.google.co.in/citations?user=X876BKcAAAAJ&hl=en&oi=sra P Please consider the environment before printing this e-mail
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
(image/png attachment: 03-image.png)
