Date: Thu, 17 Dec 2020 12:28:46 +0900
Dear All,
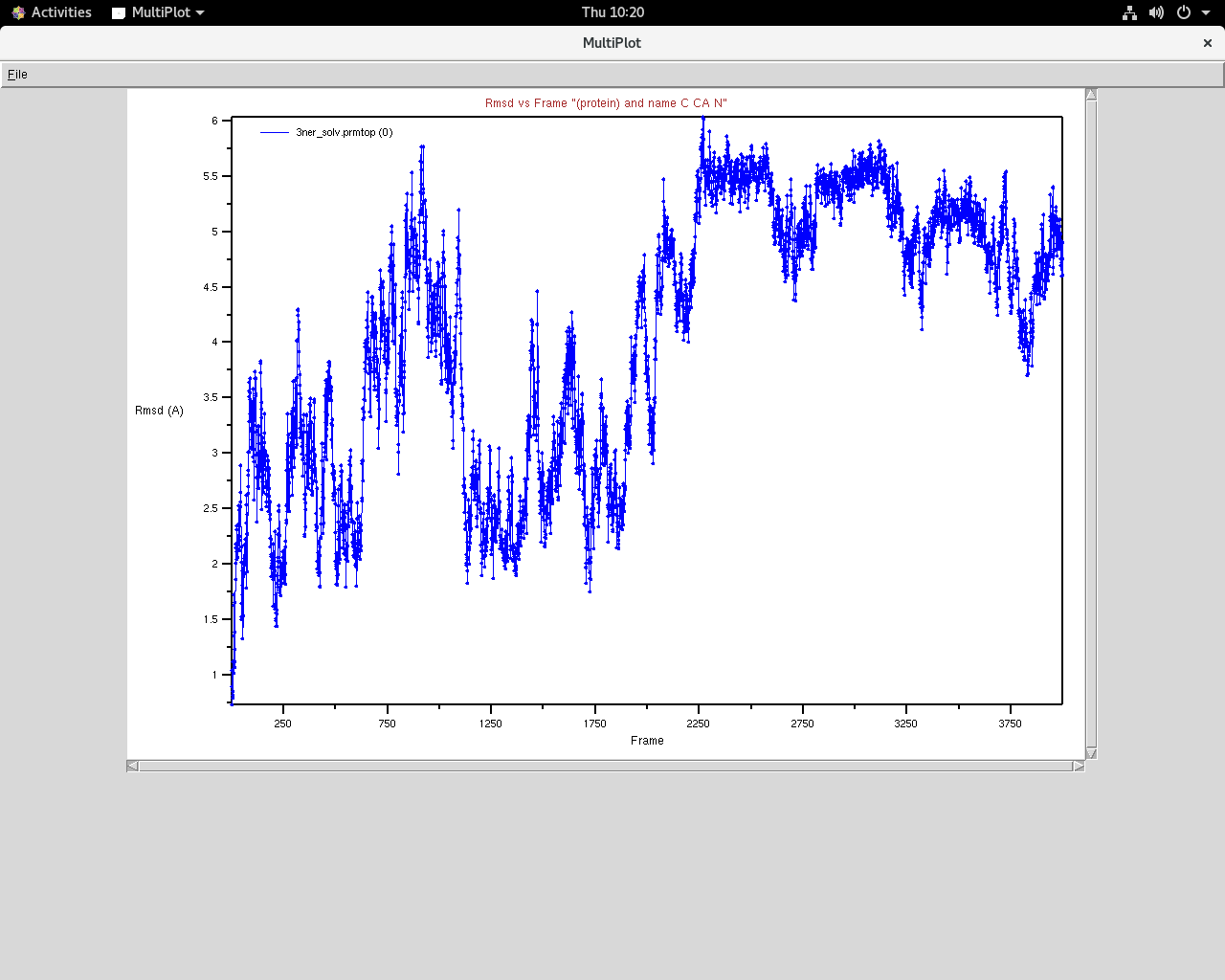
As seen in the snapshots below, the E distribution of the system is
skewed and it seems to have equilibrated only in the last 2000 frames. But
it also seems like its changing conformation which I suspect is mostly due
to the two relatively free terminals loops of the protein.
Thus can you please suggest how to ensure that the system is equilibrated
before I start extracting Es and do other types of analysis with the same?
I have also kept the number of protein atoms, solvent, and ions constant
during the system setup for the two redox states.
Thanks a lot for valuable insights into the way Amber Es are calculated.
[image: image.png]
[image: image.png]
[image: image.png]
On Thu, Dec 17, 2020 at 1:29 PM Thomas Cheatham <tec3.utah.edu> wrote:
>
> Jason's reply is excellent as usual and also accurate, despite the fact
> that AMBER is now a side job for him and he is graciously answering on his
> own time. Absolute energies have little meaning. The +/- does not matter,
> only the relative.energies.
>
> If you look at AMBER vs. CHARMM with solvated energetics, AMBER is more
> negative since CHARMM water models add a vdw on the hydrogen on their water
> (TIPS3P?) model making more vdw energy and positivity. Is it better/worse;
> look for the agreement to experiment. Yet, absolute energy values only
> matter when you can directly compare to an (exactly) equivalent system,
>
> Details matter, as do assumptions. The assumption that negative is
> "favorable" (or required) is wrong - it is all relative...
>
> We previously did direct potential energy comparisons; required equal # of
> waters, ions, etc.
>
> https://pubmed.ncbi.nlm.nih.gov/24835734/
>
> --tec3
> ________________________________________
> From: Jason Swails <jason.swails.gmail.com>
> Sent: Wednesday, December 16, 2020 8:59:35 PM
> To: AMBER Mailing List
> Subject: Re: [AMBER] positive Amber FF energies?
>
> On Wed, Dec 16, 2020 at 11:32 AM Vaibhav Dixit <vaibhavadixit.gmail.com>
> wrote:
>
> > Dear All,
> > I'm getting positive Amber FF energy values after I stripped the solvent,
> > and ions.
> > I did this for other proteins recently and got meaningful and -ve E
> values.
> > Thus I'm a bit puzzled about what these energy values might mean.
> >
>
> You've hit the nail on the head. They mean absolutely nothing. The only
> thing that has any meaning whatsoever are energy differences.
>
> Constant shifts in the potential energy surface (for example, changing bond
> potentials from [k*(x-x0)^2] to [k*(x-x0)^2 - 1e5] have no effect on forces
> and, by extension, trajectories.
>
> Also note that stripping the solvent and ions is a huge deal, and can
> completely change whether a species is stable or not. As an example, the
> dominant species in an ensemble of an isolated amino acid is its
> zwitterionic form, where the amine group is positively charged and its
> carboxylate negative. This flips in the absence of an aqueous solvent
> (which works to stabilize those charges), and the dominant stable state in
> the gas phase is its uncharged amine-carboxylic acid form.
>
> >From the printout you posted below, it doesn't seem like you turned on any
> kind of implicit solvent like GB, so I would not be at all surprised to
> learn that many configurations are only stable because of the effects of
> the solvent. But it's important to note that positive MM energies by
> themselves are completely meaningless. In fact, the values you show seem
> perfectly sensible. Bonds and angles are treated as ideal springs and so
> cannot have an energy lower than 0, and dihedrals are frequently the same
> way. The electrostatic interactions are slightly attractive, as are the
> van der Waals (again, not surprising as the vdW potential is universally
> attractive at longer distances and removing solvent and ions also removes
> many potential close contacts where a positive vdW interaction was offset
> by an attractive electrostatic one).
>
> But for a solvated system, the bulk of the net electrostatic energy comes
> from water-water interactions, which are highly favorable (and have a
> negative value by virtue of the electrostatic potential energy function).
> They don't contribute to bonded terms because their bonds are constrained
> to their minimum-energy value (0), so they basically only contribute
> nonbonded terms that tend to be negative. Ditto with the ions.
>
> The above discussion highlights why positive values make sense in this
> context. It's important to reiterate that the actual energy values in an
> MM calculation are completely meaningless.
>
> Visual inspection of the structure did not reveal anything strange and the
> > structure looks fine.
> > Thus can you please comment as to what might be wrong here and how can I
> > possibly fix this problem?
> >
> > Sample Es for stripped protein
> > NSTEP ENERGY RMS GMAX NAME NUMBER
> > 1 * 7.7864E+02 1.9139E+01* 1.1727E+02 FE
> 1491
> >
> > BOND = 284.3194 ANGLE = 828.9885 DIHED =
> > 508.5032
> > VDWAALS = -682.0879 EEL = -4975.7223 HBOND =
> > 0.0000
> > 1-4 VDW = 343.3292 1-4 EEL = 4397.5842 RESTRAINT =
> > 0.0000
> > CMAP = 73.7232
> > minimization completed, ENE= 0.77863756E+03 RMS= 0.191394E+02
> > TRAJENE: Trajectory file ended
> > TRAJENE: Trajene complete.
> >
> > Sample E for solvated protein
> > NSTEP ENERGY RMS GMAX NAME NUMBER
> > 1 * -6.1452E+04 1.3795E+01* 1.3013E+02 C
> 727
> >
> > BOND = 308.1534 ANGLE = 820.6275 DIHED =
> > 504.6346
> > VDWAALS = 8617.0697 EEL = -75800.8695 HBOND =
> > 0.0000
> > 1-4 VDW = 337.3554 1-4 EEL = 3694.9279 RESTRAINT =
> > 0.0000
> > CMAP = 66.4508
> >
>
> HTH,
> Jason
>
> --
> Jason M. Swails
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- Regards, Dr. Vaibhav A. Dixit, Visiting Scientist at the Manchester Institute of Biotechnology (MIB), The University of Manchester, 131 Princess Street, Manchester M1 7DN, UK. AND Assistant Professor, Department of Pharmacy, ▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄ Birla Institute of Technology and Sciences Pilani (BITS-Pilani), VidyaVihar Campus, street number 41, Pilani, Rajasthan 333031. India. Phone No. +91 1596 255652, Mob. No. +91-7709129400, Email: vaibhav.dixit.pilani.bits-pilani.ac.in, vaibhavadixit.gmail.com http://www.bits-pilani.ac.in/pilani/vaibhavdixit/profile https://www.linkedin.com/in/vaibhav-dixit-b1a07a39/ ORCID ID: https://orcid.org/0000-0003-4015-2941 http://scholar.google.co.in/citations?user=X876BKcAAAAJ&hl=en&oi=sra P Please consider the environment before printing this e-mail
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber

(image/png attachment: image.png)
(image/png attachment: 02-image.png)
(image/png attachment: 03-image.png)
