Date: Thu, 10 Dec 2020 15:48:17 +0530
Sorry for the inconvenience caused. I haven't attached the picture of my
last time slice as I have mentioned in the message body. I am attaching the
image here.
With regards,
Satyajit Khatua
---------- Forwarded message ---------
From: SATYAJIT KHATUA <satyajitkhatua09.gmail.com>
Date: Thu, Dec 10, 2020 at 5:38 AM
Subject: Re: [AMBER] Tri-coordination environment in zinc
To: <amber.ambermd.org>
Hi,
Have checked the para that you have suggested and the solution to overcome
that problem. Thanks a lot for pointing that out (Did not meet with such a
problem in the past. Whenever I used MCPB.py, I checked upto parmed
step for the metal site parameters).
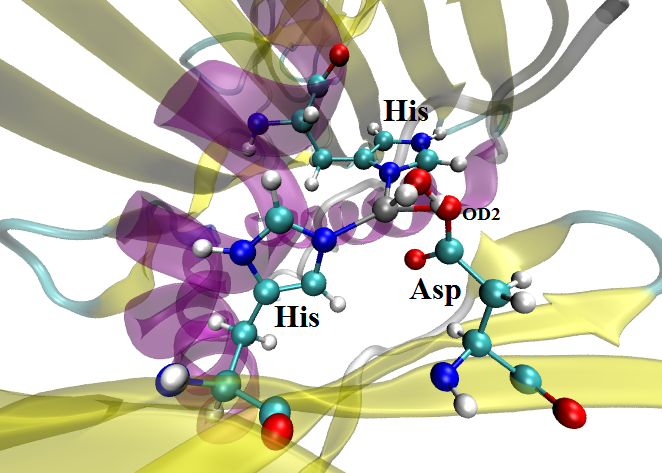
My zinc coordination site contains one ASP residue (connected to zinc
through its OD2 atom) and I think one H atom of the added water involving
some interactions with that OD2 atom of ASP (attaching the reference image
where my simulation met with the problem during heating).
Going through the solutions, I have some queries that need to be clarified.
For my case, do I have to put the harmonic restraints only on the added
water or on both the ASP residue and the added water? Do I have to treat
all the bonded residues in a nonbonded way? Your help is much appreciated.
With regards,
Satyajit Khatua
---------- Forwarded message ---------
Message: 2
Date: Mon, 7 Dec 2020 15:30:21 -0500
From: Pengfei Li <ambermailpengfei.gmail.com>
Subject: Re: [AMBER] Tri-coordination environment in zinc
To: AMBER Mailing List <amber.ambermd.org>
Message-ID: <90A7857E-C4B3-419C-AFCF-3117BE2789E5.gmail.com>
Content-Type: text/plain; charset=us-ascii
And you can find the corresponding solution in that paragraph as well.
> On Dec 7, 2020, at 3:29 PM, Pengfei Li <ambermailpengfei.gmail.com> wrote:
>
> Hi Satyajit,
>
> If using GaussView works, you do not need to use VMD to try again.
>
> Based on your description, you might meet the problem mentioned in the
second last paragraph of this tutorial:
https://ambermd.org/tutorials/advanced/tutorial20/mcpbpy.htm <
https://ambermd.org/tutorials/advanced/tutorial20/mcpbpy.htm>.
>
> Pengfei
>
>> On Nov 21, 2020, at 5:01 AM, SATYAJIT KHATUA <satyajitkhatua09.gmail.com
<mailto:satyajitkhatua09.gmail.com>> wrote:
>>
>> Hi,
>>
>> I have tried it with GaussView. First, I have created a valacy in the
zinc
>> ion and then replaced it with a water molecule. After That, I have saved
>> the coordinates into a PDB file and followed subsequent steps as
specified
>> in MCPB.py tutorial. After optimization and frequency calculation, I also
>> have a look into the fchk file to check whether there are any
>> discrepancies. But nothing suspicious is there. So I proceed further by
>> generating prmtop and inpcrd file for my MD run. I have minimized my
system
>> with SHAKE by putting a weak restraint of 2.0. After minimization, I got
>> stuck into the heating process after step 37 with the error message:
>>
>> vlimit exceeded for step 37; vmax = 76.6693
>>
>> Coordinate resetting (SHAKE) cannot be accomplished,
>> deviation is too large
>> NITER, NIT, LL, I and J are : 0 3 4651 9219 9221
>>
>> Note: This is usually a symptom of some deeper
>> problem with the energetics of the system.
>>
>> nmropt was on during my heating process. I have checked the mdcrd file by
>> saving each step for nstlim=1000. Going with the atom ids, I found out
that
>> something is going on between the O and a H atom of our newly created
water
>> molecule. I don't know how to proceed further as I have tested and tried
>> almost every suggestion that was given in the amber archive. Is there
>> anything I have done wrong?
>>
>> I will definitely try to build the system with VMD again as per your
>> suggestion. Let's see what happens.
>>
>> Thanks,
>> Satyajit Khatua
>>
>>
>> ---------- Forwarded message ---------
>> Message: 13
>> Date: Fri, 20 Nov 2020 10:42:47 -0500
>> From: Pengfei Li <ambermailpengfei.gmail.com <mailto:
ambermailpengfei.gmail.com>>
>> Subject: Re: [AMBER] Tri-coordination environment in zinc
>> To: AMBER Mailing List <amber.ambermd.org <mailto:amber.ambermd.org>>
>> Message-ID: <E475C039-5450-4525-85C7-A3BED9A61E79.gmail.com <mailto:
E475C039-5450-4525-85C7-A3BED9A61E79.gmail.com>>
>> Content-Type: text/plain; charset=us-ascii
>>
>> Hi Satyajit,
>>
>> Do you have graphic program like GaussView? GaussView can do such a job.
I
>> guess VMD could also do that by using a plugin like Molefacture:
>> https://www.ks.uiuc.edu/Research/vmd/plugins/molefacture/ <
https://www.ks.uiuc.edu/Research/vmd/plugins/molefacture/> <
>> https://www.ks.uiuc.edu/Research/vmd/plugins/molefacture/ <
https://www.ks.uiuc.edu/Research/vmd/plugins/molefacture/>>.
>>
>> After you adding the coordinated water molecules and save the coordinates
>> into a PDB file, you can just copy the coordinates of the added water
>> molecules into your original PDB file with renaming that water residue
>> (renaming both residue name and atom names inside) consistent with the
>> AMBER naming scheme (residue name: HOH or WAT, and atom names are O, H1,
>> and H2), and resequencing the PDB file using pdb4amber.
>>
>> Hope it helps,
>> Pengfei
>>
>>> On Nov 3, 2020, at 12:06 AM, SATYAJIT KHATUA <satyajitkhatua09.gmail.com
<mailto:satyajitkhatua09.gmail.com>>
>> wrote:
>>>
>>> Hi,
>>>
>>> Can you suggest to me any other tool to do so?
>>>
>>> with regards,
>>> Satyajit Khatua
>>>
>>>
>>> ---------- Forwarded message ---------
>>> Message: 6
>>> Date: Sun, 1 Nov 2020 10:35:31 -0800
>>> From: Bill Ross <ross.cgl.ucsf.edu <mailto:ross.cgl.ucsf.edu>>
>>> Subject: Re: [AMBER] Tri-coordination environment in zinc
>>> To: amber.ambermd.org <mailto:amber.ambermd.org>
>>> Message-ID: <eec483ff-eba4-ed99-e6d4-0dc151e5a5e3.cgl.ucsf.edu <mailto:
eec483ff-eba4-ed99-e6d4-0dc151e5a5e3.cgl.ucsf.edu>>
>>> Content-Type: text/plain; charset=utf-8; format=flowed
>>>
>>> Yes. It's harder to do with xleap than other graphic editors, but still
>>> might only take a few minutes.
>>>
>>> Bill
>>>
>>> On 11/1/20 9:38 AM, SATYAJIT KHATUA wrote:
>>>> Hi,
>>>>
>>>> Thanks for replying. Here, I am talking about one water intending to
>>>> satisfy the metal site coordination. Can I place the water by hand
using
>>>> the leap editor?
>>>>
>>>> with regards,
>>>> Satyajit Khatua
>>>>
>>>>
>>>> ---------- Forwarded message ---------
>>>> Message: 7
>>>> Date: Thu, 29 Oct 2020 23:59:57 -0700
>>>> From: Bill Ross <ross.cgl.ucsf.edu <mailto:ross.cgl.ucsf.edu>>
>>>> Subject: Re: [AMBER] Tri-coordination environment in zinc
>>>> To: amber.ambermd.org <mailto:amber.ambermd.org>
>>>> Message-ID: <9c0e181c-eae2-73db-90f6-a9f4970b1131.cgl.ucsf.edu <mailto:
9c0e181c-eae2-73db-90f6-a9f4970b1131.cgl.ucsf.edu>>
>>>> Content-Type: text/plain; charset=utf-8; format=flowed
>>>>
>>>> If we're just talking about a few waters, why not just place them by
>> hand?
>>>>
>>>> Bill
>>>>
>>>>
>>>> On 10/29/20 10:39 PM, SATYAJIT KHATUA wrote:
>>>>> Hi,
>>>>>
>>>>> Sorry for replying this late. I thought that maybe using MCPB.py is a
>>>>> viable solution in this case and stopped following further updates
>> posted
>>>>> in the archive. Now, I understand the problems that I am going to face
>> if
>>>> I
>>>>> don't take care of this problem that you have mentioned. Previously, I
>>>> have
>>>>> also tried the modeling by striping the crystal waters of the PDB and
>>> then
>>>>> adding the suitable water box to see if any water molecule falls
within
>>>> the
>>>>> coordination distance of zinc ion but none to avail. Can you please
help
>>>> me
>>>>> with the ways about how to saturate the metal site with the water
>>>> molecule?
>>>>> That will be helpful.
>>>>>
>>>>> with regards,
>>>>> Satyajit Khatua
>>>>>
>>>>>
>>>>> ---------- Forwarded message ---------
>>>>> Message: 7
>>>>> Date: Thu, 22 Oct 2020 17:22:53 -0400
>>>>> From: Pengfei Li <ambermailpengfei.gmail.com <mailto:
ambermailpengfei.gmail.com>>
>>>>> Subject: Re: [AMBER] Tri-coordination environment in zinc
>>>>> To: AMBER Mailing List <amber.ambermd.org <mailto:amber.ambermd.org>>
>>>>> Message-ID: <2D91EE0A-7C34-4DFF-BA87-7654944421CE.gmail.com <mailto:
2D91EE0A-7C34-4DFF-BA87-7654944421CE.gmail.com>>
>>>>> Content-Type: text/plain; charset=us-ascii
>>>>>
>>>>> Hi Satyajit,
>>>>>
>>>>> For this case maybe it is better for you to saturate the metal site
>> first
>>>>> with an additional water before the MCPB.py modeling. If the metal
site
>>> is
>>>>> not saturated, it could be challenging to get meaningful results for
the
>>>>> quantum calculations.
>>>>>
>>>>> Pengfei
>>>>>
>>>>>> On Sep 19, 2020, at 12:36 AM, SATYAJIT KHATUA <
>>> satyajitkhatua09.gmail.com <mailto:satyajitkhatua09.gmail.com>
>>>>> wrote:
>>>>>> Dear Users,
>>>>>>
>>>>>> Anyone here have worked with tri-coordination environment on zinc in
>>>>> protein?? Recently, I am working on a superantigen protein that
contains
>>> a
>>>>> tri-coordinated environment (two bound His- and one Asp-) on zinc.
>>> Usually
>>>>> zinc possess a tetra-coordination. In crystallography paper, they told
>>>> that
>>>>> possibly there is a water molecule on fourth position although they
>> didnt
>>>>> find any signature on electron density map. So can someone give some
>>>> inputs
>>>>> about the parameterization of this zinc coordination through amber
force
>>>>> fields?? Any suggestions about some tutorials or papers in this regard
>>>> will
>>>>> also be appreciated.
>>>>>> Thanks in advance,
>>>>>> Satyajit Khatua
>>>>>> _______________________________________________
>>>>>> AMBER mailing list
>>>>>> AMBER.ambermd.org <mailto:AMBER.ambermd.org>
>>>>>> http://lists.ambermd.org/mailman/listinfo/amber
>>>>> _______________________________________________
>>>>> AMBER mailing list
>>>>> AMBER.ambermd.org <mailto:AMBER.ambermd.org>
>>>>> http://lists.ambermd.org/mailman/listinfo/amber
>>>> _______________________________________________
>>>> AMBER mailing list
>>>> AMBER.ambermd.org <mailto:AMBER.ambermd.org>
>>>> http://lists.ambermd.org/mailman/listinfo/amber
>>> _______________________________________________
>>> AMBER mailing list
>>> AMBER.ambermd.org <mailto:AMBER.ambermd.org>
>>> http://lists.ambermd.org/mailman/listinfo/amber
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org <mailto:AMBER.ambermd.org>
>> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: time_slice_37.png)
