Date: Sat, 21 Nov 2020 16:35:00 +0800 (GMT+08:00)
Dear All,
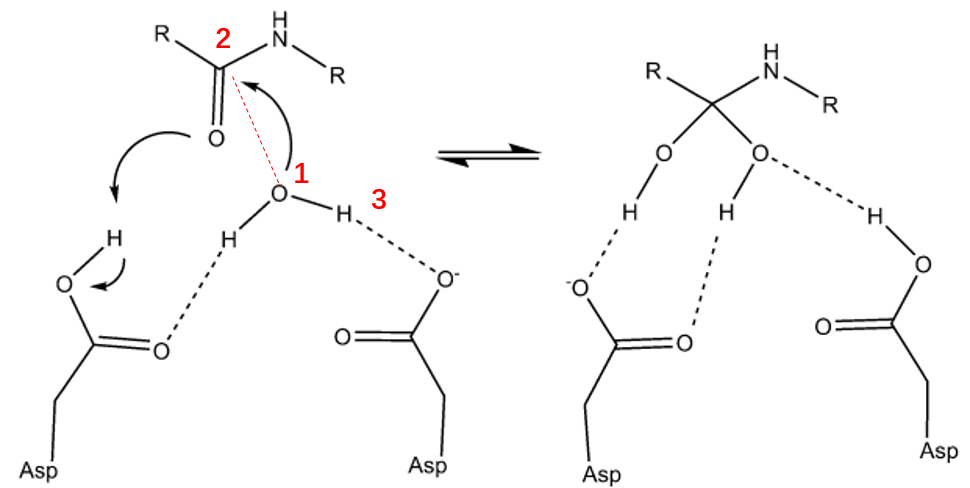
I am going to simulated a SMD using LCOD by Amber as show in the Fig,
The distance between the water oxygen (1) and the carbonyl carbon (2) of the scissile bond was adopted as the reaction coordinate,
The main parameters is as follows:
type = LCOD ! Linear Combination of Distance
i = (3,1,2,1)
r = (1.0,-1.0)
path = (distance(3,1), distance(2,1))
harm = (1000.0)
end variable
I wonder the i list and path is correct or not, any suggestion will be much appreciate.
Zhihong xin
College of Food Science and technology
Nanjing Agricultural University
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 11.png)
