Date: Thu, 19 Nov 2020 13:26:01 +0100
Hello,
I used Amber/Orca/Plumed to do QM/MM metadynamics simulations to study a
chemical reaction,
but I found that the substrate, which was treated with QM method, was
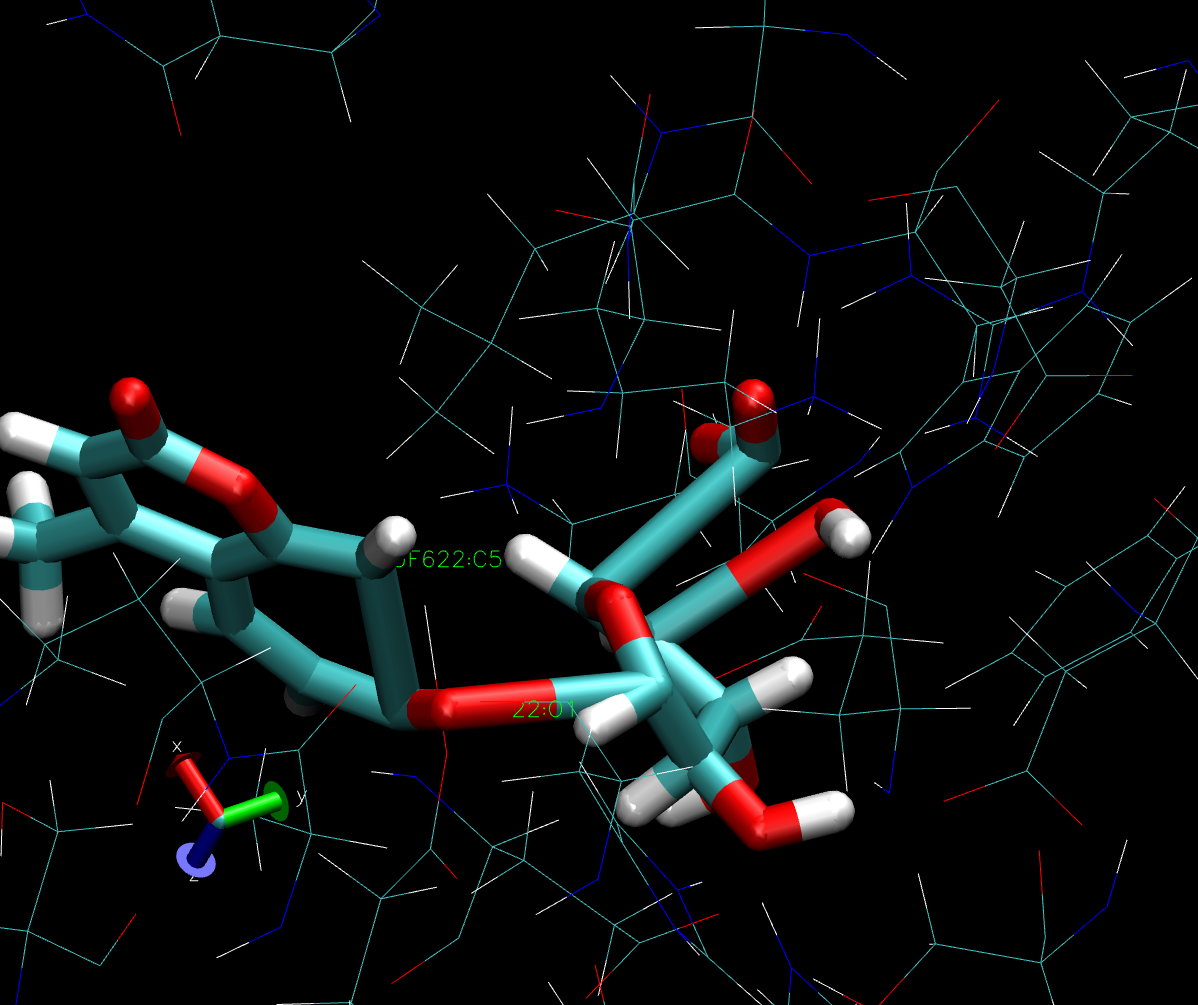
twisted unphysically, as attached.
In the attached figure, the carboxylate and hydroxyl groups on the sugar
ring are twisted, even the benzene ring are open.
But none of the bond breaking is related to the chemical reaction.
Similar problem was also observed when I used DFTB3 as QM method.
Even the artifacts happened during the simulation, the simulation was
not crashed, but went on. Does someone also have similar problem?
What is the possible reason for the unexpected issues? Any comment is
appreciated!
All the best,
Qinghua
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: vmdscene.png)
- text/plain attachment: orc_job.tpl
- text/plain attachment: md.in
