Date: Thu, 29 Oct 2020 00:47:59 -0500
Dear AMBER Users,
There is a toolkit named cellulose builder
(https://onlinelibrary.wiley.com/doi/full/10.1002/jcc.22959) which I have
used to generate the cellulose bundles. The output of this toolkit is
(.pdb) files and (.psf) file.
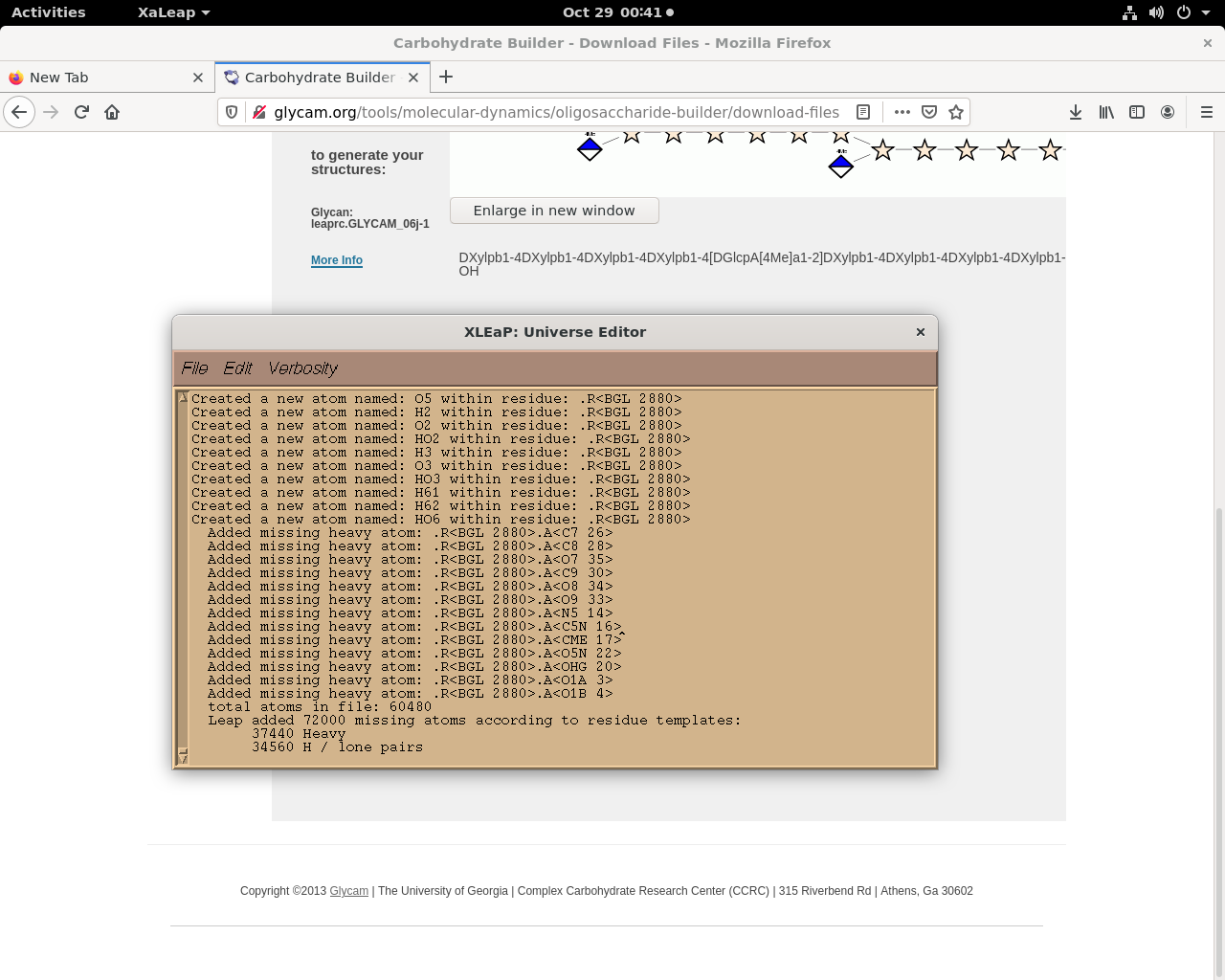
When I am importing these (.pdb) files in xleap, it gives the message
attached herewith.
*Leap added 72000 missing atoms according to residue templates:*
*37440 Heavy*
* 34560 H / lone pairs*
*The file contained 31680 atoms not in residue templates*
Could you please give me any suggestions to solve this issue?
Any help would be appreciated.
Thank you!
Regards,
-- Pinky, Sharmi AL,US
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_from_2020-10-29_00-41-23.png)
