Date: Sat, 3 Oct 2020 19:33:44 +0800
Dear AMBER Community,
I would like to ask for your help in making sense of my MMGBSA/MMPBSA
results. I separately docked a short peptide onto two receptors. I also
included calculations for an alanine scan on five of the peptide's amino
acid residues.
In my initial calculation, I used the following parameters:
&general
startframe=1, endframe=100000, interval=100, verbose=1,
ligand_mask=":1761-1783", receptor_mask=":1-1760",
strip_mask=":WAT,PA,PC,PE,OL,K+,Cl-,CHL",
# entropy=1,
/
&gb
igb=2, saltcon=0.150,
/
&pb
istrng=0.100,
memopt=1,
inp=2, radiopt=0,
/
&alanine_scanning
/
However, MMPBSA results showed positive binding energy values. In another
thread in this mailing list that featured a similar issue, it was suggested
that inp=1 should be used instead of inp=2 to avoid obtaining positive
values in MMPBSA. I reran my calculations using inp=1 and obtained the
following results.
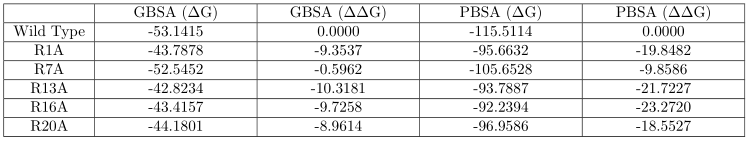
receptor A:
[image: image.png]
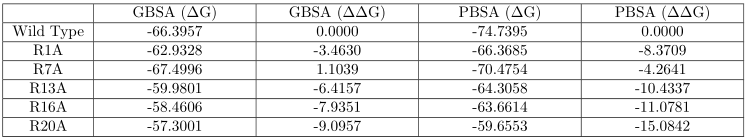
receptor B:
[image: image.png]
Here are my questions:
(1) There is a massive disparity between the MMGBSA and MMPBSA results on
the potassium channel. Is this normal? Aren't the MMGBSA and MMPBSA results
supposed to be fairly similar in magnitude since one is simply an
approximation of another?
(2) Are the results on different receptors comparable? Is it safe to say
that according to the MMGBSA results, the peptide should prefer binding to
receptor B rather than receptor A? But this is contradicted by the MMPBSA
results.
If these questions are deemed trivial, I will greatly appreciate being
recommended the right papers to read.
Best,
Rashid
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
