Date: Thu, 17 Sep 2020 16:16:19 +0530
Dear Amber users,
I would like to mention you that I am doing a cmd simulation of a protein
containing Br atom in it and would like to study its impact on the
structure. Also, would like to study presence of other halogen group such
as Cl and I in the same pocket where Br is present in the crystal.
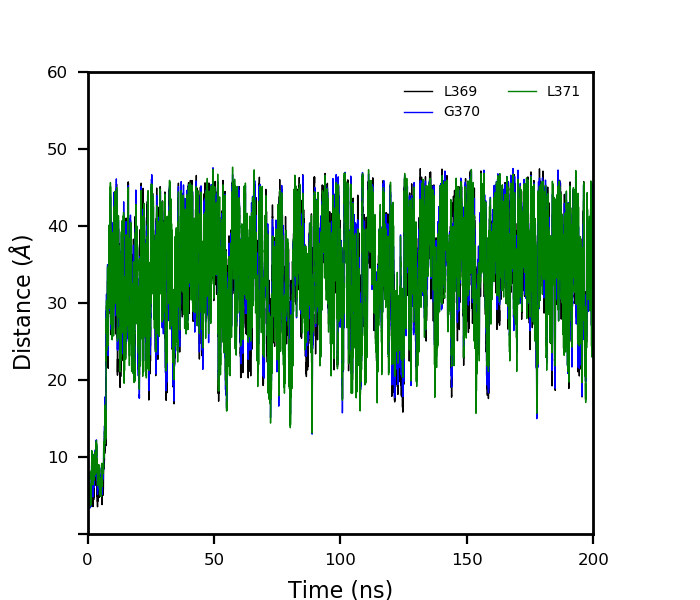
It is reported that some of the protein's residue interact with the Br or
Cl. So I performed the MD simulation and checked the interaction distance
throughout the trajectory.
But soon the ions moved out of the pocket.
How can I proceed to study such interactions? What forcefield should I
choose for I (iodide)?
The interaction plot of the same I am attaching here.
Any help or suggestions are highly appreciated.
Thanks and regards
-- Nisha Amarnath Jonniya PhD Research Scholar Biosciences and Biomedical Engineering Indian Institute of Technology, Indore India
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: distance-crystal-4q2a.png)
