Date: Mon, 6 Jul 2020 12:46:41 -0500
Apologies for the multiple emails. I had just realized that I did not
attach the respective file. It is now attached herein.
P.S. While I have tried to unwrap and autoimage, there is a chance that I
may have not utilized these action commands correctly. Any and all
advice would be greatly appreciated.
Sincerely,
*An T. Ta *
Department of Chemistry | Ph.D. Candidate
Coolbaugh Hall | Room 118
Golden, CO 80401
On Mon, Jul 6, 2020 at 12:44 PM An Ta <anta.mymail.mines.edu> wrote:
> Dear AMBER users and colleagues,
>
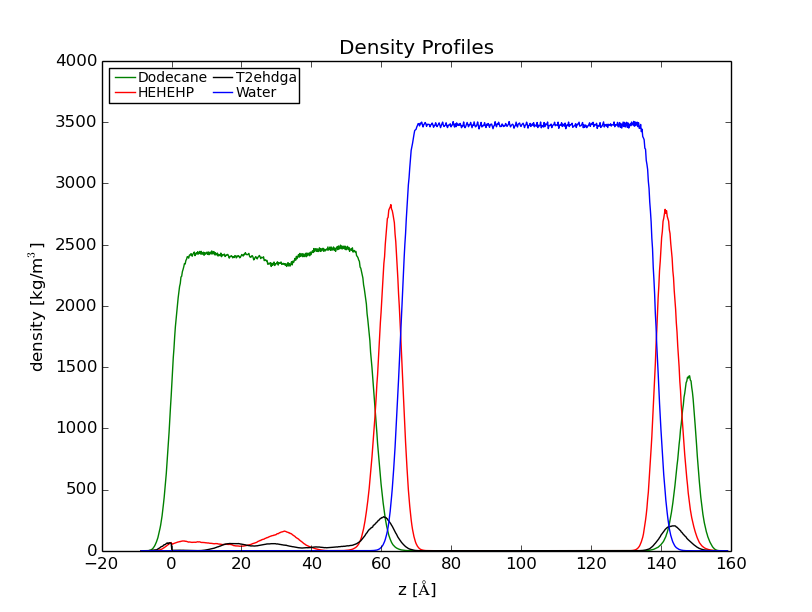
> I have been attempting to correct multiple density profiles for a biphasic
> system. The simulations were performed with PBC on a system that contains a
> water-dodecane solvent and two specific surfactants. Currently, I am trying
> to account for PBC in the density analyses since I am observing "erroneous"
> peaks at the edge of my box. The density profiles were calculated such that
> the mass density of my constituents are assessed in reference to the
> z-dimension of my box. I have tried the unwrap and autoimage action
> commands and the profiles still possess the erroneous peaks at the edge of
> the box.
>
> I have included one of the density profiles that I am trying to correct
> for in this thread. This specific plot was calculated with a trajectory
> that has neither been unwrapped or autoimaged. Even so, density plots
> calculated with the new trajectories that were unwrapped or autoimaged
> looked similar to the one provided. Thank you for your consideration and
> any advisement.
>
> Sincerely,
> *An T. Ta *
> Department of Chemistry | Ph.D. Candidate
> Coolbaugh Hall | Room 118
> Golden, CO 80401
>
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: DensProfile_0M1_SecondRun.png)
