Date: Sun, 26 Apr 2020 22:59:07 -0500
Thank you.
*Query 1: Regarding lignin:*
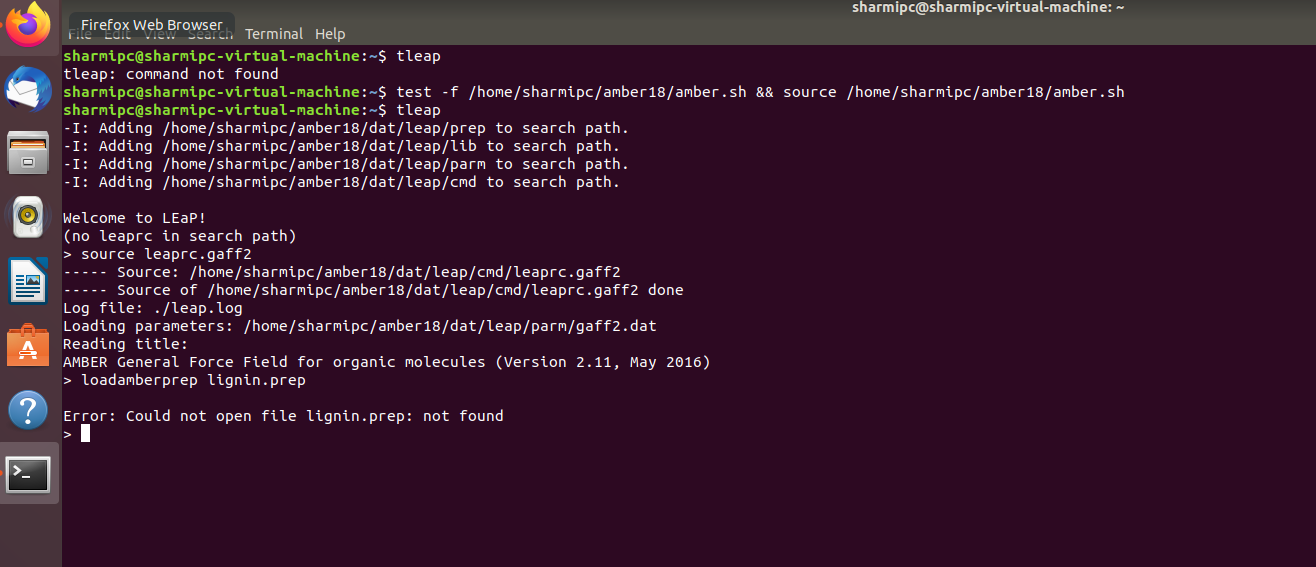
But when I have tried according to your instructions, it says *could not
open the file: lignin.prep not found*. I have attached the screenshot
(li.png) of that.
I am using Amber 18 licensed version with Amber2019 Tools.
*Query 2 : Regarding cellulose-hemicellulose chain: *
To make the interaction between cellulose-hemicellulose chain, I need to
place them closer and parallel to each other.
But I have used this command in xleap:
source leaprc.GLYCAM_06j-1
cellulose = loadpdb cellulose.pdb
hemicellulose = loadpdb hemicellulose.pdb
sum = combine { cellulose hemicellulose }
edit sum
saveamberparm sum parm7 rst7
savepdb sum.pdb
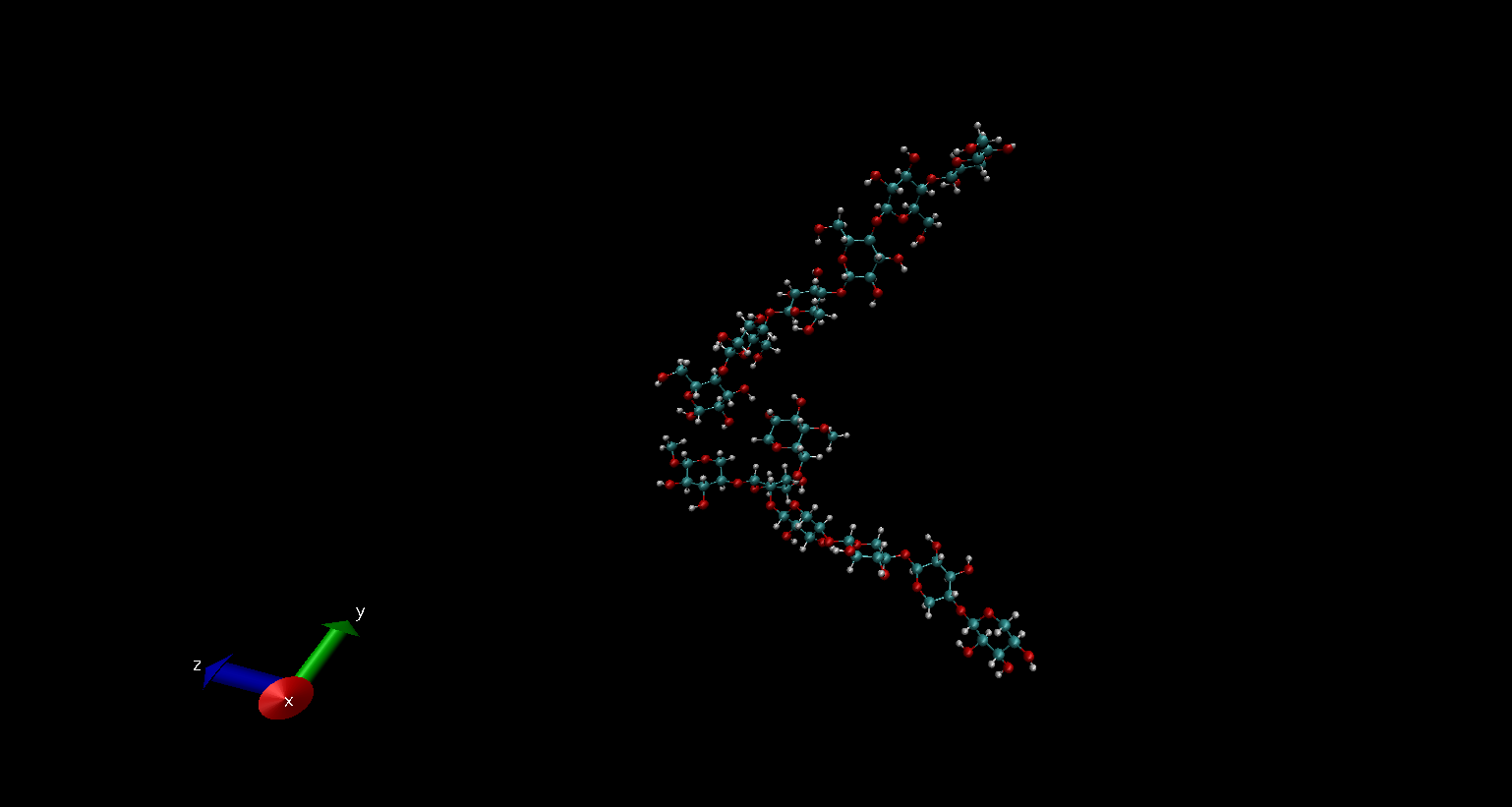
and I found this the chains are connected like this. Please find the
attached image (cell_hemi interaction) herewith. However, I need to place
them close enough to see the hydrogen bonds between them. Similarly, I Need
to do this for cellulose bundles too. To do this what commands or procedure
I need to follow? Please let me know.
And if I want to relax the system of each chain *(individually or connected
together),* what minimization input file should I use? Is it similar like
Tutorial B0?
Thank you for your patience reading.
I will be waiting for your response.
Thank you again.
Sincerely,
Pinky
On Fri, Apr 24, 2020 at 6:37 AM Lachele Foley <lf.list.gmail.com> wrote:
> You're going to need to generate a prep file for the molecule.
> Antechamber can do that. The manual explains reasonably well, but ask
> if you have difficulty.
>
> Then, in leap:
>
> source leaprc.gaff2
> loadamberprep lignin.prep
> lignin = loadpdb lignin.pdb
>
> On Fri, Apr 24, 2020 at 3:30 AM Pinky Mazumder <pmazumder67.gmail.com>
> wrote:
> >
> > Hi Lachele,
> >
> > Hope you are well.
> >
> > I have generated a pdb file of another polymer (lignin) using graphic
> antechamber. Then, if I want to load it into xleap, what command should I
> use?
> >
> >
> > Thank you.
> >
> > Sincerely yours,
> >
> > Pinky
> >
> > On Wed, Apr 22, 2020, 7:17 AM Lachele Foley <lf.list.gmail.com> wrote:
> >>
> >> Don't use pdb4amber. it is already in AMBER format. You also do not
> >> need to use reduce.
> >>
> >> You will need to alter the charge as described at the link I posted
> >> above. You must add the charge modification to the leap commands I
> >> gave above.
> >>
> >> On Mon, Apr 20, 2020 at 9:30 PM Pinky Mazumder <pmazumder67.gmail.com>
> wrote:
> >> >
> >> > Hi Lachele,
> >> >
> >> > I have converted the pdb files using the following command:
> >> >
> >> > pdb4amber -i orig.pdb -o new.pdb --reduce --dry
> >> >
> >> >
> >> > However, I have loaded the pdb files in xleap which is edited by you
> and have checked that if the total charge are zero or not. I found that the
> unit hema_TER.pdb is not zero.
> >> >
> >> > Attached is the screenshot of this. Please find this.
> >> >
> >> >
> >> > How do I solve this?
> >> >
> >> > Sincerely,
> >> > Pinky
> >> >
> >> > On Mon, Apr 20, 2020 at 1:48 AM Lachele Foley <lf.list.gmail.com>
> wrote:
> >> >>
> >> >> What do you mean by "converted"? Do you mean that you added TER
> cards
> >> >> to the PDB file? Did you do anything else?
> >> >>
> >> >> The way to really check bonding is to generate topology/coordinate
> >> >> files (parm7/rst7) and then load them into a viewer like VMD or
> >> >> Chimera. When I do that, and output a set of pamr7/rst7 files, the
> >> >> bonding looks ok. I don't have xleap at easy access at the moment,
> so
> >> >> I'm can't check the behavior there, but it's not super important.
> >> >>
> >> >> Note that you will also need to alter the charge on the C atom where
> >> >> the MEX is attached. The website does that automatically where it
> >> >> can, but there is no standard way to encode charge in a PDB file, so
> >> >> if you need to use the PDB file like you are, then you will need to
> >> >> adjust charge. The doc is below.
> >> >>
> >> >>
> http://glycam.org/docs/help/2014/04/04/adding-chemical-derivatives-to-glycam-residues/
> >> >>
> >> >> I used these commands in tleap:
> >> >>
> >> >> source leaprc.GLYCAM_06j-1
> >> >> m = loadpdb hema_TER.pdb ## I added TER cards to the PDB file you
> >> >> sent a couple emails ago
> >> >> check m ## this gets tleap to perform some simple checks on your
> >> >> molecule, including total charge - should be zero if correct
> >> >> saveamberparm m m.parm7 m.rst7
> >> >> quit
> >> >>
> >> >> Then, I loaded them into vmd with:
> >> >>
> >> >> vmd m.parm7 m.rst7
> >> >>
> >> >> Make sure that this works by itself, then try to combine this and
> your
> >> >> other molecule.
> >> >>
> >> >> I've attached the PDB file with the TER cards added.
> >> >>
> >> >> Let me know if you have other questions.
> >> >>
> >> >> On Mon, Apr 20, 2020 at 1:05 AM Pinky Mazumder <
> pmazumder67.gmail.com> wrote:
> >> >> >
> >> >> > Hi Lachele,
> >> >> >
> >> >> > Hope you are doing well.
> >> >> >
> >> >> > I am facing another problem. I have build the hemicellulose chain
> using glycam carbo builder. so, for loading it in xleap, it needed to be
> converted.
> >> >> >
> >> >> >
> >> >> > The converted pdb looks fine in vmd. However, when I have loaded
> it into xleap, I found that the alpha-glucoronic residue have detached from
> the beta-xylose chains.
> >> >> >
> >> >> >
> >> >> > Attached is the image of vmd as well as xleap. Could you please
> tell me why this is happening?
> >> >> >
> >> >> >
> >> >> > Thank you.
> >> >> >
> >> >> > Sincerely,
> >> >> > pinky
> >> >> >
> >> >> > On Sat, Apr 18, 2020 at 2:21 AM Lachele Foley <lf.list.gmail.com>
> wrote:
> >> >> >>
> >> >> >> Sorry I didn't reply before now. Did you figure it out? Do you
> still
> >> >> >> need help?
> >> >> >>
> >> >> >> On Fri, Apr 17, 2020 at 5:28 PM Pinky Mazumder <
> pmazumder67.gmail.com> wrote:
> >> >> >> >
> >> >> >> > Hi Lachele,
> >> >> >> >
> >> >> >> > As you said about the combine command. It works.
> >> >> >> >
> >> >> >> > Thank you so much.
> >> >> >> >
> >> >> >> > Sincerely,
> >> >> >> > Pinky
> >> >> >> >
> >> >> >> > On Fri, Apr 17, 2020 at 9:59 AM Pinky Mazumder <
> pmazumder67.gmail.com> wrote:
> >> >> >> >>
> >> >> >> >> Also I do not have the Amber style pdb file. Do I need this
> for combining the two pdb?
> >> >> >> >>
> >> >> >> >> Thank you.
> >> >> >> >>
> >> >> >> >> Sincerely,
> >> >> >> >> Pinky
> >> >> >> >>
> >> >> >> >> On Fri, Apr 17, 2020 at 2:45 AM Pinky Mazumder <
> pmazumder67.gmail.com> wrote:
> >> >> >> >>>
> >> >> >> >>> Thank you for your cordial response.
> >> >> >> >>>
> >> >> >> >>> Here I have attached the two pdb files of cellulose and
> hemicellulose.
> >> >> >> >>>
> >> >> >> >>> I need to combine those chain as a single pdb so that I can
> run the relaxation.
> >> >> >> >>>
> >> >> >> >>>
> >> >> >> >>> Thank you again.
> >> >> >> >>>
> >> >> >> >>> Sincerely,
> >> >> >> >>> Pinky
> >> >> >> >>>
> >> >> >> >>> On Fri, Apr 17, 2020 at 1:06 AM Lachele Foley <
> lf.list.gmail.com> wrote:
> >> >> >> >>>>
> >> >> >> >>>> That usually means you have a bonding issue. You will often
> need to
> >> >> >> >>>> make little edits to your PDB file and/or leap commands to
> get carbs
> >> >> >> >>>> to be loaded properly.
> >> >> >> >>>>
> >> >> >> >>>> Did you download the amber-style pdb file? Can you send me
> your pdb file?
> >> >> >> >>>>
> >> >> >> >>>> On Fri, Apr 17, 2020 at 1:59 AM Pinky Mazumder <
> pmazumder67.gmail.com> wrote:
> >> >> >> >>>> >
> >> >> >> >>>> > Hi Lachele,
> >> >> >> >>>> >
> >> >> >> >>>> > Thank you for your reply.
> >> >> >> >>>> >
> >> >> >> >>>> >
> >> >> >> >>>> > I have build the hemicellulose using glycam carbohydrate
> builder and then I
> >> >> >> >>>> > am trying to load the pdb file of ''hemicellulose'' using
> loadpdb command
> >> >> >> >>>> > in xleap.
> >> >> >> >>>> >
> >> >> >> >>>> > But, It shows the error !FATAL: Message: Atom named C1
> from 4LA did not
> >> >> >> >>>> > match !
> >> >> >> >>>> >
> >> >> >> >>>> > Attached is the screenshot.
> >> >> >> >>>> >
> >> >> >> >>>> > Do you have any idea why this is happening?
> >> >> >> >>>> >
> >> >> >> >>>> > Any help would be appreciated.
> >> >> >> >>>> >
> >> >> >> >>>> >
> >> >> >> >>>> > Thank you again.
> >> >> >> >>>> >
> >> >> >> >>>> >
> >> >> >> >>>> > Sincerely,
> >> >> >> >>>> > Pinky
> >> >> >> >>>> >
> >> >> >> >>>> > On Thu, Apr 16, 2020 at 3:23 AM Lachele Foley <
> lf.list.gmail.com> wrote:
> >> >> >> >>>> >
> >> >> >> >>>> > > I usually use tleap rather than xleap, but you should be
> able to use
> >> >> >> >>>> > > either. See my previous reply, but also consider the
> 'combine'
> >> >> >> >>>> > > command.
> >> >> >> >>>> > >
> >> >> >> >>>> > > Learning to use xleap or tleap takes a little time, but
> the Amber
> >> >> >> >>>> > > manual does a pretty good job of describing the
> functions.
> >> >> >> >>>> > >
> >> >> >> >>>> > > On Wed, Apr 15, 2020 at 11:14 PM Pinky Mazumder <
> pmazumder67.gmail.com>
> >> >> >> >>>> > > wrote:
> >> >> >> >>>> > > >
> >> >> >> >>>> > > > Dear Amber Users,
> >> >> >> >>>> > > >
> >> >> >> >>>> > > > I have built cellulose chain using xleap command. On
> the other hand,
> >> >> >> >>>> > > > I have made the chain of hemicellulose using GLYCAM_06
> carbohydrate
> >> >> >> >>>> > > builder.
> >> >> >> >>>> > > >
> >> >> >> >>>> > > > Now, I need to put the chain close together.
> >> >> >> >>>> > > >
> >> >> >> >>>> > > >
> >> >> >> >>>> > > > So, my question is, how can I make the both chains
> closer together?
> >> >> >> >>>> > > Should
> >> >> >> >>>> > > > I load the chain of hemicellulose in xleap? If it is,
> then what is the
> >> >> >> >>>> > > > command for this job?
> >> >> >> >>>> > > >
> >> >> >> >>>> > > >
> >> >> >> >>>> > > > Any help would be appreciated. Please forgive me if I
> am asking something
> >> >> >> >>>> > > > silly.
> >> >> >> >>>> > > >
> >> >> >> >>>> > > > Thank you.
> >> >> >> >>>> > > >
> >> >> >> >>>> > > > --
> >> >> >> >>>> > > > Pinky
> >> >> >> >>>> > > > AL,US
> >> >> >> >>>> > > > _______________________________________________
> >> >> >> >>>> > > > AMBER mailing list
> >> >> >> >>>> > > > AMBER.ambermd.org
> >> >> >> >>>> > > > http://lists.ambermd.org/mailman/listinfo/amber
> >> >> >> >>>> > >
> >> >> >> >>>> > >
> >> >> >> >>>> > >
> >> >> >> >>>> > > --
> >> >> >> >>>> > > :-) Lachele
> >> >> >> >>>> > > Lachele Foley
> >> >> >> >>>> > > CCRC/UGA
> >> >> >> >>>> > > Athens, GA USA
> >> >> >> >>>> > >
> >> >> >> >>>> > > _______________________________________________
> >> >> >> >>>> > > AMBER mailing list
> >> >> >> >>>> > > AMBER.ambermd.org
> >> >> >> >>>> > > http://lists.ambermd.org/mailman/listinfo/amber
> >> >> >> >>>> > >
> >> >> >> >>>> >
> >> >> >> >>>> >
> >> >> >> >>>> > --
> >> >> >> >>>> > Pinky
> >> >> >> >>>> > AL,US
> >> >> >> >>>> > _______________________________________________
> >> >> >> >>>> > AMBER mailing list
> >> >> >> >>>> > AMBER.ambermd.org
> >> >> >> >>>> > http://lists.ambermd.org/mailman/listinfo/amber
> >> >> >> >>>>
> >> >> >> >>>>
> >> >> >> >>>>
> >> >> >> >>>> --
> >> >> >> >>>> :-) Lachele
> >> >> >> >>>> Lachele Foley
> >> >> >> >>>> CCRC/UGA
> >> >> >> >>>> Athens, GA USA
> >> >> >> >>>>
> >> >> >> >>>> _______________________________________________
> >> >> >> >>>> AMBER mailing list
> >> >> >> >>>> AMBER.ambermd.org
> >> >> >> >>>> http://lists.ambermd.org/mailman/listinfo/amber
> >> >> >> >>>
> >> >> >> >>>
> >> >> >> >>>
> >> >> >> >>> --
> >> >> >> >>> Pinky
> >> >> >> >>> AL,US
> >> >> >> >>
> >> >> >> >>
> >> >> >> >>
> >> >> >> >> --
> >> >> >> >> Pinky, Sharmi
> >> >> >> >> AL,US
> >> >> >> >
> >> >> >> >
> >> >> >> >
> >> >> >> > --
> >> >> >> > Pinky, Sharmi
> >> >> >> > AL,US
> >> >> >>
> >> >> >>
> >> >> >>
> >> >> >> --
> >> >> >> :-) Lachele
> >> >> >> Lachele Foley
> >> >> >> CCRC/UGA
> >> >> >> Athens, GA USA
> >> >> >
> >> >> >
> >> >> >
> >> >> > --
> >> >> > Pinky, Sharmi
> >> >> > AL,US
> >> >>
> >> >>
> >> >>
> >> >> --
> >> >> :-) Lachele
> >> >> Lachele Foley
> >> >> CCRC/UGA
> >> >> Athens, GA USA
> >> >> _______________________________________________
> >> >> AMBER mailing list
> >> >> AMBER.ambermd.org
> >> >> http://lists.ambermd.org/mailman/listinfo/amber
> >> >
> >> >
> >> >
> >> > --
> >> > Pinky, Sharmi
> >> > AL,US
> >>
> >>
> >>
> >> --
> >> :-) Lachele
> >> Lachele Foley
> >> CCRC/UGA
> >> Athens, GA USA
>
>
>
> --
> :-) Lachele
> Lachele Foley
> CCRC/UGA
> Athens, GA USA
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- Pinky, Sharmi AL,US
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: li.PNG)
(image/png attachment: cell_hem_interaction.PNG)
