Date: Sun, 26 Apr 2020 10:19:45 +0530
> Dear all,
>The following is a recurring problem and I have repeated the simulation
multiple times and still the issue persists. Please help.
I have done a simulation on an aromatic ligand which must remain planar. I
optimized the structure in Gaussian, done docking on the ligand, calculated
ESP charges for selected conformers in Gaussian again. My ligand has a +2
charge. Then I have done the RESP fitting using antechamber and generated
the frcmod file.(AMBER18)
> antechamber -fi gout -i ligand.log -fo prepi -o ligand.prepi -c resp
> parmchk2 -i ligand.prepi -o ligand.frcmod -f prepi.
>
> Then I performed restrained minimization, heating and density
equilibration and unrestrained equilibration and 500ns production. My
ligand strangely loses the planarity by the equilibration stage and shows a
really distorted structure which is unacceptable. I have used the gaff ff
and Tip3p waterbox in the simulation. My frcmod file looks like the
following:
> DIHE
> ca-ca-cc-cc 4 2.800 180.000 2.000 same as X
-c2-ca-X , penalty score=232.0
> ca-ca-cc-cf 4 2.800 180.000 2.000 same as X
-c2-ca-X , penalty score=232.0
> ca-cc-cf-ha 4 16.000 180.000 2.000 same as X
-cc-cd-X , penalty score=136.0
> ca-cc-cf-cf 4 16.000 180.000 2.000 same as X
-cc-cd-X , penalty score=136.0
> cc-cc-cf-ha 4 16.000 180.000 2.000 same as X
-cc-cd-X , penalty score=136.0
> cc-cc-cf-cf 4 16.000 180.000 2.000 same as X
-cc-cd-X , penalty score=136.0
>
> IMPROPER
> c3-ca-na-cc 1.1 180.0 2.0 Using the
default value
> ca-ca-ca-na 1.1 180.0 2.0 Using the
default value
> ca-ca-ca-cc 1.1 180.0 2.0 Using the
default value
> ca-ca-ca-ha 1.1 180.0 2.0 Using general
improper torsional angle X- X-ca-ha, penalty score= 6.0)
> cc-h4-cc-na 1.1 180.0 2.0 Using the
default value
> ca-cc-cc-cf 1.1 180.0 2.0 Using the
default value
> cc-cf-cf-ha 1.1 180.0 2.0 Same as X -X
-ca-ha, penalty score= 46.8 (use general term))
> c3-ca-na-cd 1.1 180.0 2.0 Using the
default value
> cc-h4-cd-na 1.1 180.0 2.0 Using the
default value
> cc-cd-cc-ha 1.1 180.0 2.0 Same as X -X
-ca-ha, penalty score= 38.9 (use general term))
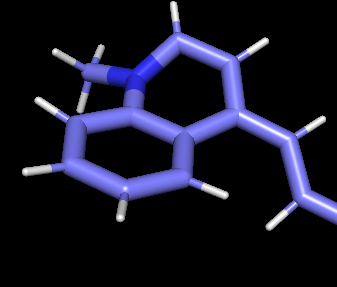
> Could someone suggest what might have went wrong and how to solve this
issue. I am attaching the image of my distorted ligand here.
> My two major doubts are
> 1. Is the frcmod file wrong?
> 2. should I include the -nc flag in the resp fitting since my ligand has
a +2 charge?
When I repeated the procedure to an old ligand which we had then simulated
in AMBER14, the newly generated frcmod files have same components as the
old files,but with a high penalty score. Is there something peculiar we
should mention in the latest version?
> Thanks in advance
>
> --
> Jenny R.S
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: nnnnn.PNG)
