Date: Tue, 14 Apr 2020 05:59:00 +0000
Hello,
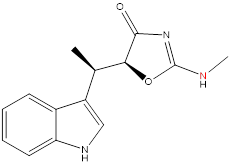
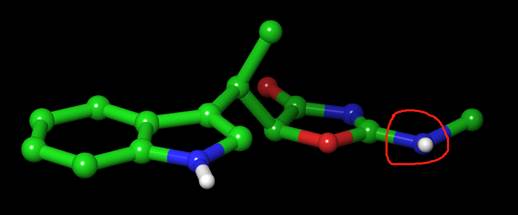
I tried a simulation with a ligand like, the red N should be sp2, so the amino group should be a plane, but during the simulation, I extract one snapshot, it was not a plane, why?
[cid:image003.png.01D61264.9306BC10][cid:image006.jpg.01D61264.9306BC10]
I prepared the ligand with command line: antechamber -i ligand.pdb -fi pdb -o ligand.mol2 -fo mol2 -c bcc; parmchk2 -i ligand.mol2 -f mol2 -o ligand.frcmod -a y
Do you have any idea why the amino was not a plane?
Thank you for the help!
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image003.png)
(image/jpeg attachment: image006.jpg)
