Date: Tue, 24 Mar 2020 13:56:28 +0100
Dear Amber Users,
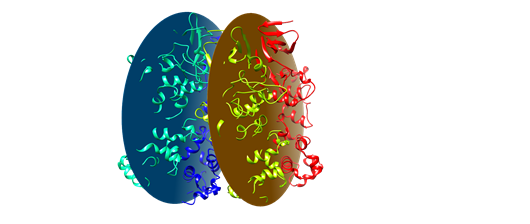
I performed an angle analysis between two dimer interface defined as any
pair of Cα atoms (example system below)
[image: image.png]
I performed this analysis with four different systems.
Since the systems investigated are rotated to the right and rotated to the
left I was expecting as output (a) positive and (b) negative angles (*i.e.*
for example + 25-30 degrees for right hand and - 25-30 for left hand
rotated)
Do you have any idea why the value received as output is always positive?
Thank you for the help!
The command line used was:
“
#Add Modules, etc etc
module load amber/16.at17
$AMBERHOME/bin/cpptraj
# load prmtop and trajectory
parm crystal_3FBV_ion_ok.prmtop
trajin reeim.xtc
#analysis with residue at the interface identify by using Chimera (i.e.
using select > ZONE)
rms first :1-1687.CA
vector A corrplane
:6,12-14-22,25-30,33-79,83-86,106-137,154-216,222-243,255-258-287-320-322,341-343-366-369,395-466-469,477-534,547-586,596-611-615,628-630-683,684-686-688,700-720-732-735-823,840.CA
vector B corrplane
:888,896-903-957,966-1005,1016-1018-1031-1034-1036,1047-1050-1103,1104-1106-1108,1120-1140-1143,1144-1150,1152-1243,1260-1261,1265-1273,1279-1281-1289,1293-1295,1299-1346,1350-1353,1373-1375-1404,1422-1484,1489-1510,1522-1559,1554-1587-1589,1607-1610-1616-1633,1634-1636,1662.CA
vectormath vec1 A vec2 B dotangle out angle_tetramer_YEAST_1.dat
run
quit
“
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
