Date: Wed, 29 Jan 2020 13:57:18 -0500
Hi all,
I am trying to follow the MCPB.py tutorial here
<http://ambermd.org/tutorials/advanced/tutorial20/mcpbpy.htm>, and apply
the procedure to a protein of interest in my group here. The tutorial
itself is very clear and well written (thanks!). however, the question
arises when trying to apply to my system.
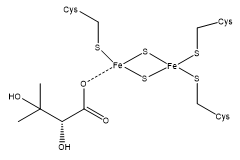
The point is that I’m trying to parametrize a 2F-2S system, where the Fe
atoms are connected to the protein by 3 Cysteines, leaving the 4th valence
open for a ligand, like this:
[image: image.png]
My question is: in the tutorial, the metal atoms get separated in one
residue for each metal atom, this residue containing just this metal atom,
by itself. This is straightforward in the case of one metal atom connected
to protein residues. However, how should I consider it here? Should I
separate each Fe atom in its own residue? And in this case, how to deal
with the S atoms? (I already parametrized the ligand.)
I could not find examples with similar sites. I found this article here
<https://doi.org/10.1016/j.bbagen.2017.05.005>, but unfortunately they
don’t provide any details on how the site was prepared, only mention that
they used MCPB… (Yes, I looked into the supplemental information… nothing
there either L)
Thanks a lot for any help you can give on this issue!
All the best,
-- Gustavo Seabra.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
