Date: Wed, 8 Jan 2020 12:25:11 -0500
Hello!
I have found that the pure solvent simulations needs to be
well-equilibrated. I have done the following:
1. Initial minimization with 500 steps steepest descent/500steps conjugate
gradient
2. Heating to 300K at constant volume over 100 ps, Berendsen thermostat.
3. Minimization with 500 steps steepest descent/500steps conjugate gradient
4. Constant pressure equilibration at 300 K and 1 atm with a coupling
constant=0.2 (again, Berendsen) for 500 ps.
5. Constant volume equilibration at 300 K for 500 ps with Berendsen.
The pure solvent simulation should have *more* atoms than the solvent
contribution of your system (solute + solvent).
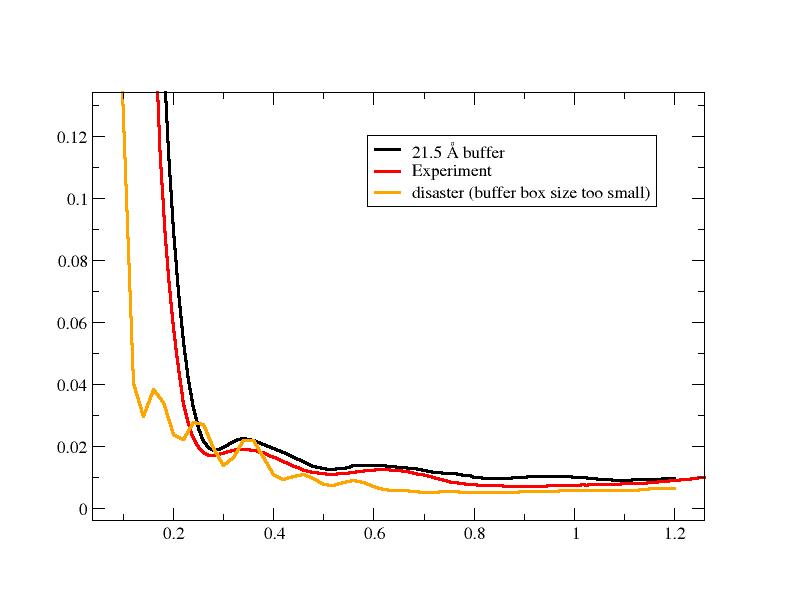
That is, I have found that the volume of pure solvent (buffer) should be >
volume of solvent + solute (sample) - you can see in the attached plot what
happens when the buffer box size is too small.
I check volumes at each step to make sure of this (can be done using the
volume command in Cpptraj),
One caveat is that the calculated spectra are only 'guaranteed' out to
small angles of q (Hung Nguyen's paper should have more information about
this:
https://aip-scitation-org.proxy-um.researchport.umd.edu/doi/pdf/10.1063/1.4953037?class=pdf
).
Good luck!
-Christina
[image: Plot.new.jpg]
On Wed, Jan 8, 2020 at 11:49 AM Buteler,Maria del Pilar <
pbuteler.chem.ufl.edu> wrote:
> Hi, I would like to calculate the saxs profile for my simulation and I'm
> trying to set up the calculation. I would like to use saxs_md and I'm
> aware I need the trajectory in pdb format of my system and also a
> simulation of pure solvent. However, should the solvent simulation have
> the same number of atoms as my original simulation and also is just
> relaxing the solvent enough? My runs are using Tip3P water.
>
> Thanks!!
>
>
>
> Pilar Buteler
>
> PhD Candidate, Roitberg Research Group
>
> Department of Chemistry, University of Florida
>
> Gainesville, FL 32611
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- ----------------------------------------------------------------- Christina Bergonzo Research Chemist Biomolecular Measurement Division, MML, NIST -----------------------------------------------------------------
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: Plot.new.jpg)
