Date: Tue, 25 Jun 2019 17:29:33 +0100
Dear Martis
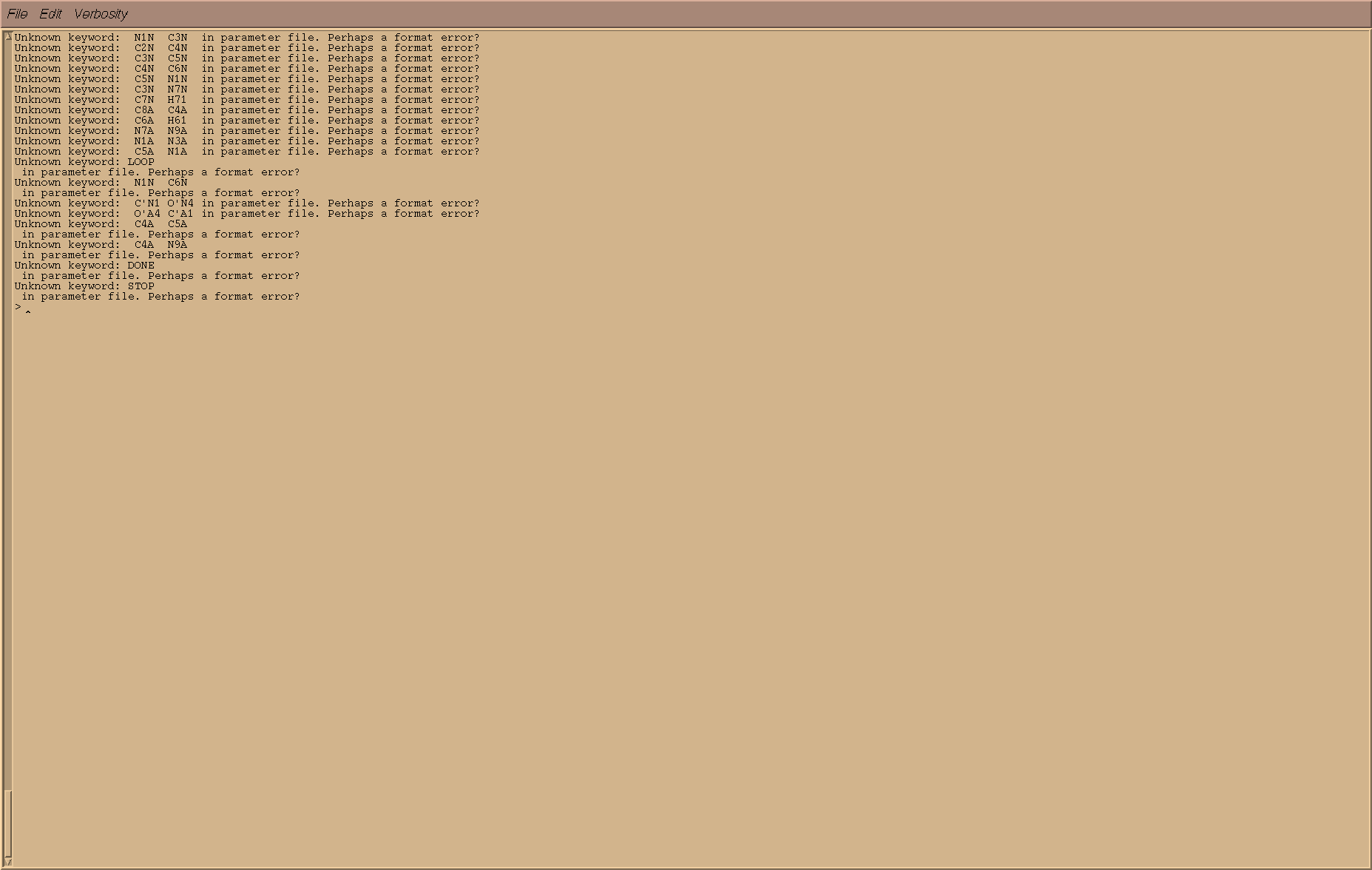
I loaded frcmod & prepi files by using following commands:
mods loadAmberParams NADP.frcmod
mods2 loadAmberParams NADP.prepi
it shows error as mentioed in screenshot.
Kindly suggest me the right way
thank you
Sadaf
On Tue, Jun 25, 2019 at 6:11 AM Elvis Martis <elvis.martis.bcp.edu.in>
wrote:
> Hello,
> you really don't need to obtain the mol2 file this way.
> I would suggest loading the frcmod and prepi files that you obtained from
> the Manchester database in xleap.
> Then see the residue name and atom names for NADP in the database and
> according modify them in your PDB file (coordinates as complexed with the
> protein). Once all atoms names and residue names are as per the prepi
> file, leap should not throw any errors.
>
> Hope this helps
>
>
> Best Regards
>
> Elvis Martis
>
>
>
> ________________________________
> From: Sadaf Rani <sadafrani6.gmail.com>
> Sent: 25 June 2019 01:55
> To: AMBER Mailing List
> Subject: Re: [AMBER] open valance problem
>
> Dear Elvis
>
> I looked for the NADP parameters as you suggested but I want to get the
> mol2 file from the same process to loads in to t leap for parameter
> generation
> as commands below:-
>
> source leaprc.protein.ff14SB
> source leaprc.gaff2
> source leaprc.water.tip3p
>
> ligA = loadmol2 NAP_amber.mol2
> ligB = loadmol2 BG6_amber.mol2
>
> loadamberparams NAP_amber.frcmod
> loadamberparams BG6_amber.frcmod
>
>
> protein = loadPdb "Combined_nolig.pdb"
> complex = combine {protein ligA ligB}
> protein = createUnit Combined_nolig.pdb
> setBox protein "vdw"
> solvateBox protein TIP3PBOX 14.0 iso
> addIonsRand complex Na+ 0
> saveamberparm complex complex.prmtop complex.inpcrd
> quit
>
>
> but when I try
>
> antechamber -i NADP_1.pdb -fi pdb -o NADP_1.mol2 -fo mol2 -c bcc -pf yes
> -nc -3 -m 1 -at gaff2 -j 4
>
> it gives error
>
> Welcome to antechamber: molecular input file processor.
>
> acdoctor mode is on: check and diagnosis problems in the input file.
> -- Check Format for pdb File --
> Status: pass
> -- Check Unusual Elements --
> Status: pass
> -- Check Open Valences --
> Status: pass
> -- Check Geometry --
> for those bonded
> for those not bonded
> Status: pass
> -- Check Weird Bonds --
> Status: pass
> -- Check Number of Units --
> Status: pass
> acdoctor mode has completed checking the input file.
>
> Info: Bond types are assigned for valence state (1) with penalty (1).
> Info: Total number of electrons: 384; net charge: -3
>
> Running: /home/srania/Amber/amber16/bin/sqm -O -i sqm.in -o sqm.out
> /home/srania/Amber/amber16/bin/to_be_dispatched/antechamber: Fatal Error!
> Cannot properly run "/home/srania/Amber/amber16/bin/sqm -O -i sqm.in -o
> sqm.out".
>
> How can I solve this error
> How can I calculate the parameters for NADP+ if I want to learn at my own
>
>
> will be really thankful for answer
>
> thank you
> Sadaf
>
>
> On Sun, Jun 23, 2019 at 4:57 AM Elvis Martis <elvis.martis.bcp.edu.in>
> wrote:
>
> > Hello
> > NADP parameters are already available
> > http://research.bmh.manchester.ac.uk/bryce/amber/.
> >
> >
> > Best Regards
> >
> > Elvis Martis
> >
> >
> >
> > ________________________________
> > From: Sadaf Rani <sadafrani6.gmail.com>
> > Sent: 22 June 2019 18:56
> > To: AMBER Mailing List
> > Subject: [AMBER] open valance problem
> >
> > Dear Amber
> > I am trying to find to parameters for co-factor NADP+ by antechamber
> > But when I use this command:-
> > antechamber -i NADP_noH.mol2 -fi mol2 -o NADP_amber.mol2 -fo mol2 -c bcc
> > -pf yes -nc -3 -at gaff2 -j 4
> >
> > It gives me this error
> >
> > Welcome to antechamber: molecular input file processor.
> >
> > acdoctor mode is on: check and diagnosis problems in the input file.
> > -- Check Format for mol2 File --
> > Status: pass
> > -- Check Unusual Elements --
> > Status: pass
> > -- Check Open Valences --
> > Warning: The number of bonds (1) for atom (ID: 33, Name: O1N) does not
> > match
> > the connectivity (2) for atom type (O.3) defined in
> > CORR_NAME_TYPE.DAT.
> > Warning: The number of bonds (1) for atom (ID: 34, Name: O1X) does not
> > match
> > the connectivity (2) for atom type (O.3) defined in
> > CORR_NAME_TYPE.DAT.
> > Warning: The number of bonds (1) for atom (ID: 39, Name: O2A) does not
> > match
> > the connectivity (2) for atom type (O.3) defined in
> > CORR_NAME_TYPE.DAT.
> > Warning: The number of bonds (1) for atom (ID: 43, Name: O2X) does not
> > match
> > the connectivity (2) for atom type (O.3) defined in
> > CORR_NAME_TYPE.DAT.
> > Warning: The number of bonds (3) for atom (ID: 55, Name: C4N) does not
> > match
> > the connectivity (4) for atom type (C.3) defined in
> > CORR_NAME_TYPE.DAT.
> > Warning: The number of bonds (3) for atom (ID: 72, Name: N9A) does not
> > match
> > the connectivity (2) for atom type (N.ar) defined in
> > CORR_NAME_TYPE.DAT.
> > But, you may safely ignore the warnings if your molecule
> > uses atom names or element names as atom types.
> > -- Check Geometry --
> > for those bonded
> > for those not bonded
> > Status: pass
> > -- Check Weird Bonds --
> > /home/srania/Amber/amber16/bin/to_be_dispatched/antechamber: Fatal Error!
> > Weird atomic valence (3) for atom (ID: 55, Name: C4N).
> > Possible open valence.
> >
> > Hoe to solve this issue
> > I have attached .mol2 file if there is something wrong please tell me.
> >
> > thank you in advance
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_from_2019-06-25_17:18:28.png)
