Date: Mon, 24 Jun 2019 13:07:33 +0200
Dear Amber people,
I am working on a system containing of 2 chains in explicit TIP3P
solvent, and I want to restrain the solute for subsequent GIST
calculations. However, the density of the NpT equilibrated system is
significantly different depending on the restraint_wt setting.
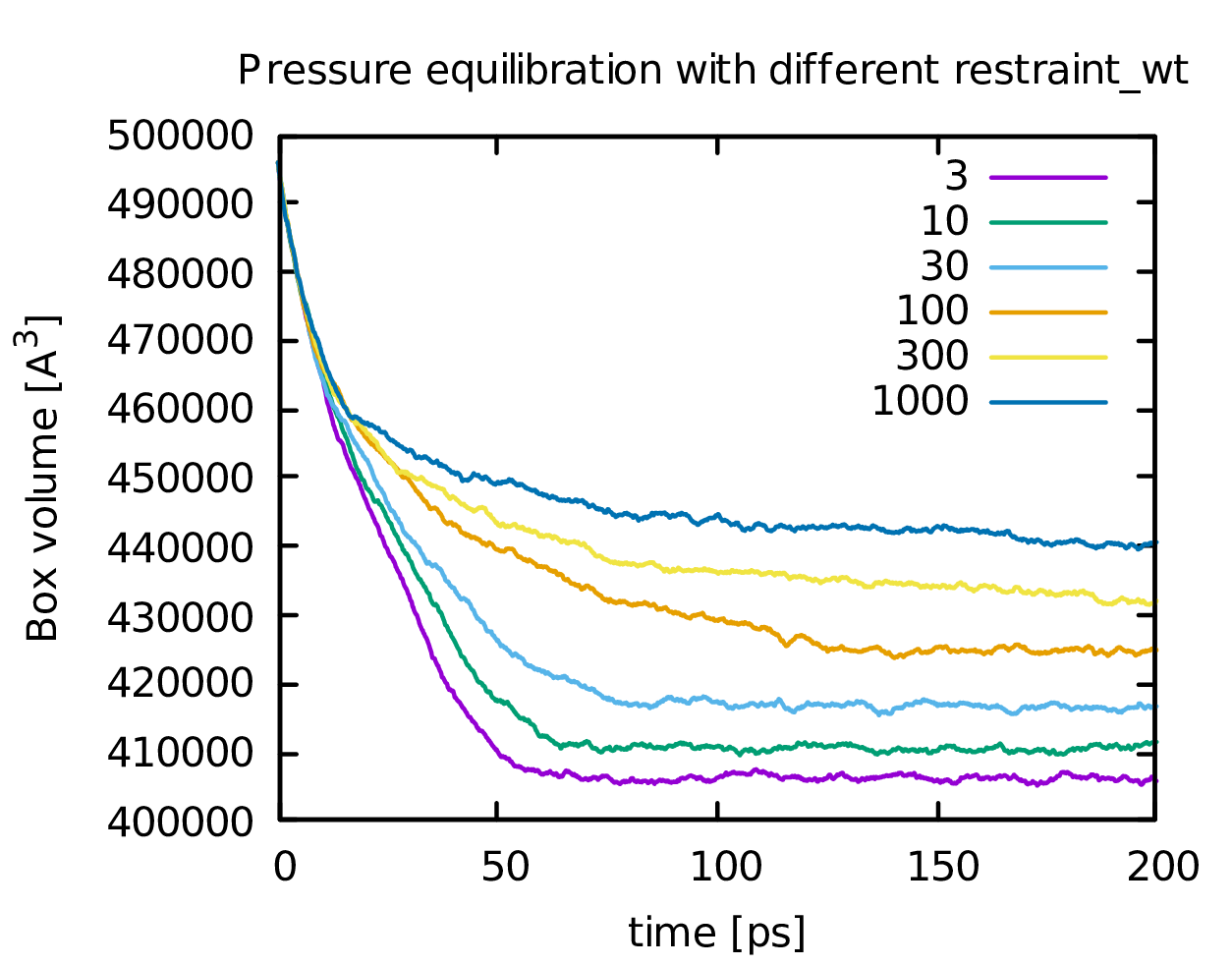
I attach a plot of the box volume vs. time with different restraint_wt
settings to illustrate the problem. I am aware that restraints of 300 or
1000 are way too strong, but the effect is already quite pronounced with
10 kcal/mol/A².
It seems to me that this is the same problem that was described in
http://archive.ambermd.org/201103/0111.html a few years ago. In brief,
the problem seems to occur because the center of mass of the 2 molecules
is scaled with the box size during pressure equilibration. This leads to
clashes between them. Of course, those clashes are larger when stronger
restraints are used. It seems that the restraint forces are not included
in the virial, but the clashes are, so that the calculated pressure of
the system is too high, and the barostat tries to counteract this by
increasing the box size again.
Is there any elegant way to avoid this problem? There seems to have been
no real solution in the original thread. A quick fix would be to do
multiple equilibration steps where each restart file is the reference
for the next step. But first of all I would still be worried about the
ensemble, and also there could be larger deviations from the starting
structure with each step. What is the recommended way of dealing with this?
Best regards,
Franz Waibl
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: box_volume_vs_time.png)
