Date: Tue, 14 May 2019 01:16:59 +0000
Hello all,
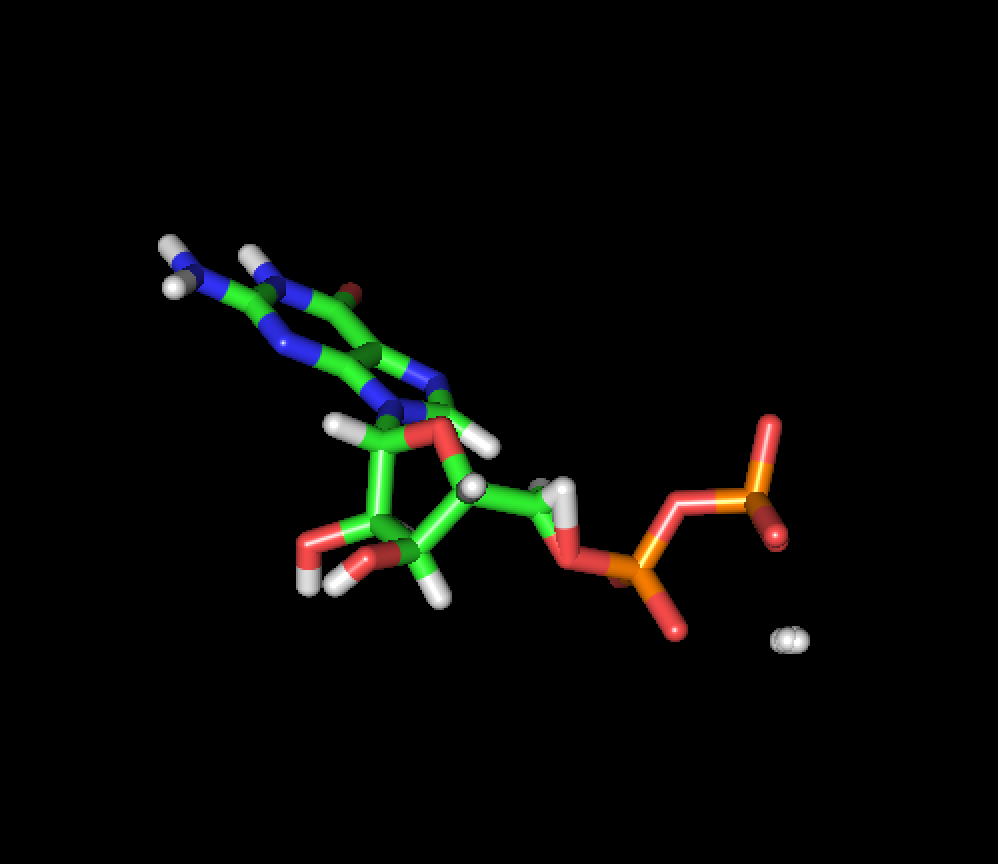
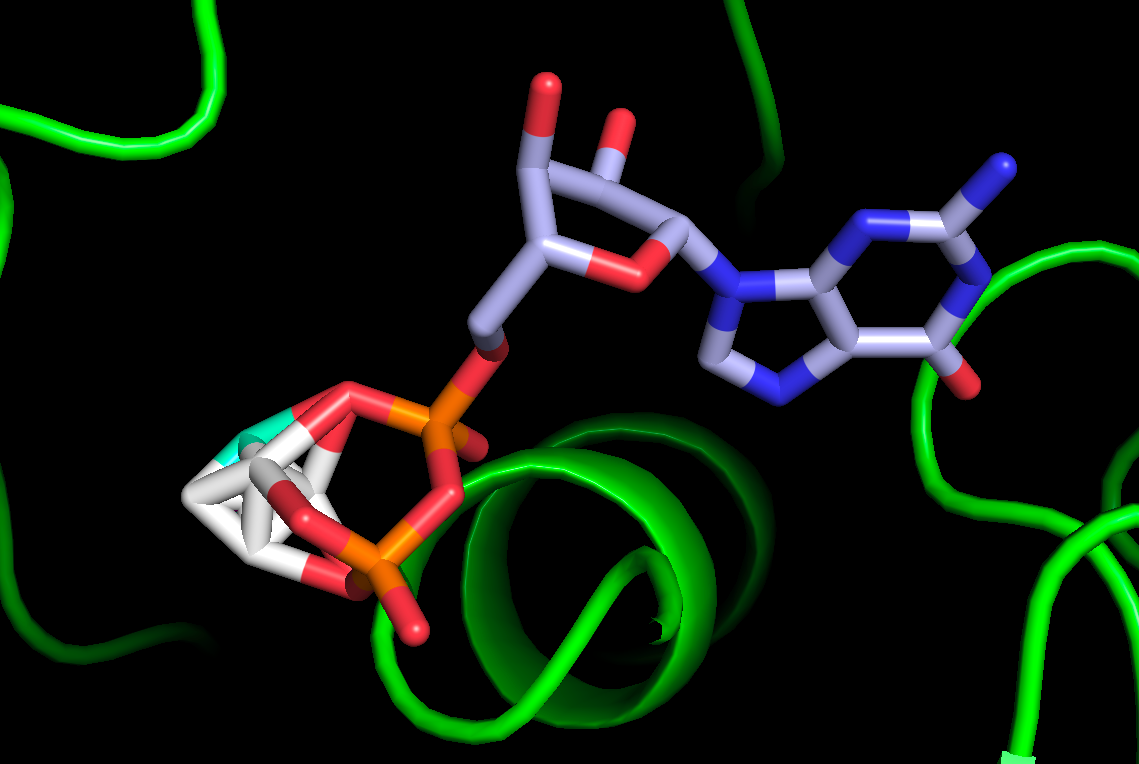
My protein contains GDP and Mg2+. After the simulation, I look up the individual frames and the average structure over the 50 ns. I found in the individual frames the Mg2+ stays close to the phosophates of GDP and leads to the artificial bonds recognized by the PyMol. But in the average structure, the distance looks fine and no artificial bonds by PyMol. I attached the screenshot below. This is something with the processing?
The parameters used for Mg is:
Oelschlaeger et al JMB 2007 Mg dummy and MD6.
When I generate the final trajectory, I used the unwrap as:
unwrap :1-169
center :1-169 mass origin
image origin center familiar
and the way I generate the average structure is:
cpptraj ../proteinA.top <<eof
trajin ./proteinA.mdcrd 25000 50000 1
rms first
average proteinA.avg.pdb pdb
=====
The average structure
[image.png]
individual frames:
[cid:a43a94a8-7a83-41c2-ba22-e46fdddcbf82]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
