Date: Mon, 13 May 2019 14:41:42 +0200 (CEST)
Dear AMBERs,
i'm hoping you're having a nice day , and wish you could help someone in
need.
So i try to make a MD on a protein with a lipid bilayer structure (created
with CHARMM-GUI). my lipid is only with DOPC
using charmmlipid4amber, to create a file that will change my DOPC into OL
and PC.
I succed to create the prmtop file and inpcrd . but after simulation i saw
that my
lipid are still split into three residues and that my bilayer structure is
slowly looking like a sphere when the head of the lipid will just float
around with water. I know that in force field lipid14 they split DOPC into
those 3 residue.
but i would like to know if there is a way to maintain those residue
together for my simulation.
i already tryed some things :
-scripting my files to recreate the bond between them, by deleting TER
when it's need it (but then tleap will split them again)
-rename my lipid in to OL , but then the ATOM (from PC) doesn't have a
type in tleap
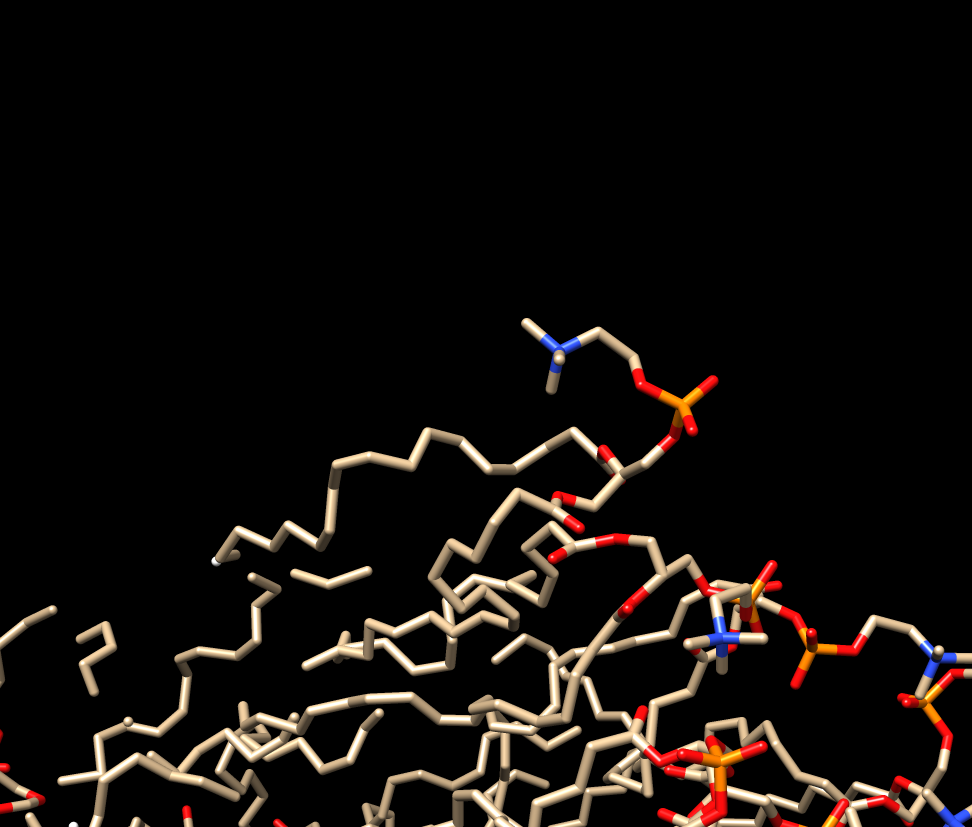
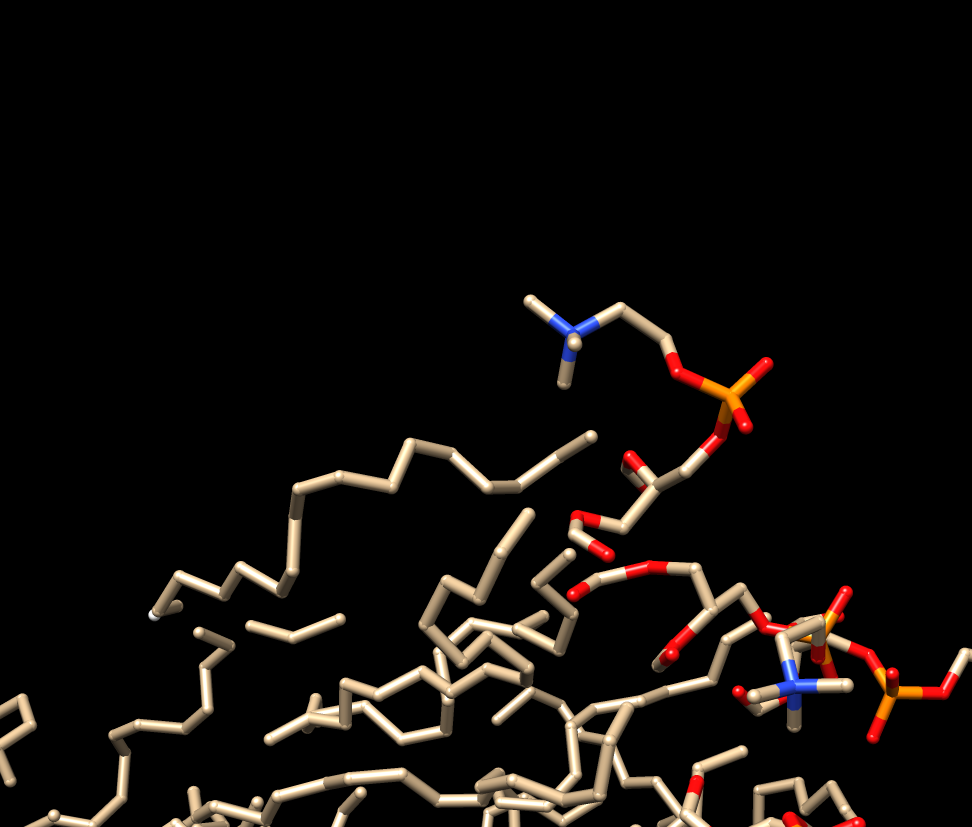
I join this with 2 images. First one from the lipid as it should be
(linked together) , and the second one when load into tleap (then using
savepdb to open it in chimera again) .
I'm greatfull that you spend some times helping me.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: imagelink.png)
(image/png attachment: imageunlink.png)
