Date: Mon, 13 May 2019 01:29:33 +0000
Hello
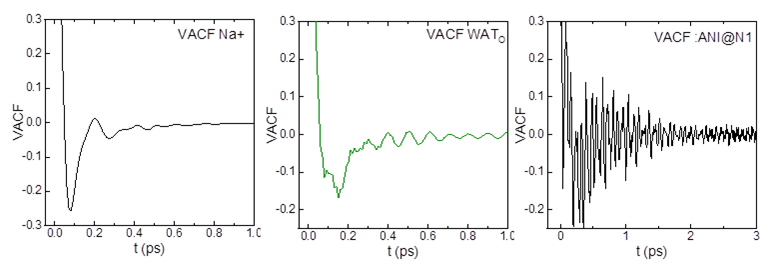
In an aqueous solution I am trying to compare the VACF of anions, cations and WAT. For the alkali cation and WAT I obtain more or less what I would expect from literature. For the anions (10 atoms) however I get really noisy curves (see figure). I ran 1ns NVT runs with dt 0.2fs recording data every 10fs. My VACF-input:
trajin ***.mdcrd mdvel ***.mdvel
velocityautocorr anion :ANI usevelocity out VACF-anion.dat diffout VACF-anion-diff.dat tstep 0.01 norm
go
The anion contains some 10 atoms and also if I select a single atom (e.g. :anion.N1) I get really noisy data. From ionic liquid literature where also rather big ions are being used I only know much smoother VACF as for e.g. alkali cations. Is there a way to calculate the VACF for the center of mass of the molecule defined in the mask? Or could it be that published VACFs are just heavily smoothened..?
Also, if I record velocities on that timescale (every 10fs), it is ok to put usevelocity, correct?
Thank you very much and best regards
David
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Clipboard02.png)
