Date: Tue, 7 May 2019 20:46:45 +0200
Dear all,
I'm coming again to this question since I have made some progress on my
system. Thank you very much, Zachary, for the script! I got this nice graph
with it!
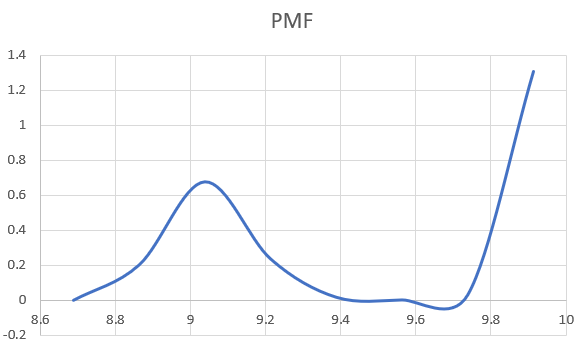
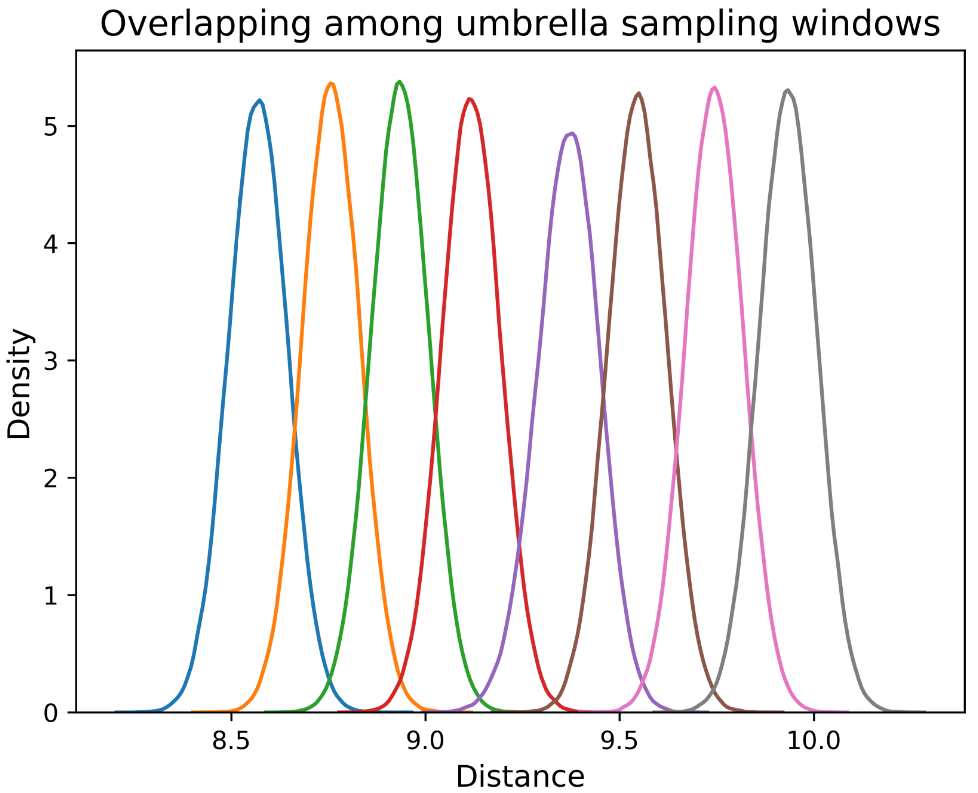
[image: image.png][image: image.png]
Density overlap of distances and the final PMF are shown.
To refresh context on my system, it is the simulation of an unbinding
process among a long peptide (20residues approx.) and a protein binding
site.
Quite a lot of interactions are given between the peptide and protein, so I
would expect to have many of them broken and formed during the unbinding
process, possibly explaining several peaks in the final PMF.
These are the new results with windows of 20ns of simulation (each), this
time using fixed r1 to 2 A and r4 to 60, starting from an 8.6 A distance,
50kcal/mol of restraint force.
Periodicity of the PMF has disappeared and seems to make more sense.
However, some questions are still unsolved...
I am still getting these 0.0000 and sudden jumps of F values on the WHAM
output (calculation done with 50(kcal/mol)·2 and 10^-6 tolerance value).
Data of the first 8 windows is shown below.
My suspicion is that it may still not be enough simulation time, since I
have another test done with 15ns per window, with a PMF which follows the
tendency of this one but with some differences in the profile.
I also suspect from the restraint force used, although I have tested lower
energies and none of them is able to keep the distance reasonably steady.
Another thing I have noticed is that all PMF plots I obtain start at 0
kcal/mol, while examples given in the tutorial and other publications
don't. I don't know if this is some type of rescaling option done after
WHAM calculation.
Any ideas to explain and correct these issues are really appreciated
#Number of windows = 8
#Iteration 10: 0.003204
#Iteration 20: 0.000417
#Iteration 30: 0.000059
#Iteration 40: 0.000008
#Iteration 50: 0.000001
#Iteration 60: 0.000000
#Coor Free +/- Prob +/-
8.687500 0.000000 0.004658 0.174297 0.000140
8.862500 0.202048 0.000446 0.124194 0.000264
9.037500 0.676175 0.000908 0.056067 0.000260
9.212500 0.235641 0.012488 0.117390 0.000174
9.387500 0.017193 0.008444 0.169343 0.000093
9.562500 0.002032 0.008927 0.173704 0.000101
9.737500 0.030852 0.002295 0.165507 0.000226
9.912500 1.305848 0.000371 0.019498 0.000236
#Window Free +/-
#0 0.000000 0.000000
#1 0.000000 0.000000
#2 1.521030 0.000483
#3 0.000000 0.000000
#4 0.000000 0.000000
#5 0.000000 0.000000
#6 0.000000 0.000000
#7 2.701136 0.000369
Thank you all very much,
Daniel
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
