Date: Sun, 28 Apr 2019 23:09:41 +0300
Hi amber lovers,
I am trying to generate new force field parameters based on quantum
mechanical calculation. I used amber manual "parameter development" title
for this and I calculated bond-angle-non bond paramters but I am having a
hard time obtaning dihedral parameters for aromatics Let's consider about
pyrazin. These are gaff2 pyrazin parameters :
ca-ca-nb-ca 2 9.600 180.000 2.000
h4-ca-nb-ca 2 9.600 180.000 2.000
nb-ca-ca-nb 4 14.500 180.000 2.000
h4-ca-ca-nb 4 14.500 180.000 2.000
h4-ca-ca-h4 4 14.500 180.000 2.000
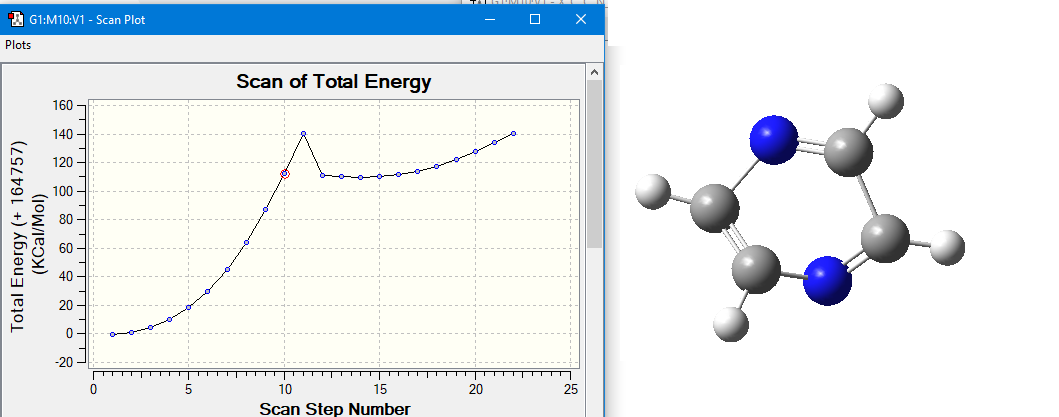
When I scanned "ca-ca-nb-ca" dihedral angels, I found that dihedral bond
is breaking at 100 degre and 111 kcal/mol energy. According to manual, if
this is an ordinary dihedral ca-ca-nb-ca should have (111/2=)55,5
kcal/mol V/2 value.
But in gaff2 it is just 9.60kcal/mol. I couldn't understand how? and why?
Beside, idivf shouln't be 4 instead of 2?
Thank you.
NOTE: Here is the picture.every step is degree.
[image: image.png]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
