Date: Mon, 1 Apr 2019 10:28:55 +0800 (GMT+08:00)
Dear all,
Here are the steps I took to build the central fragment of a modified nucleotide in the AMBER force fields. I was wondering if you could read these steps and tell me what is wrong or what can I do to simplify the process.
Steps:
1.Charge Derivation for the modified nucleotide.
1.1 optimize geometry for dimethylphosphate(DMP) and modified nucleoside
input files: DMP_OPT.gjf mG_OPT.gjf
output files: DMP_OPT.out mG_OPT.out
1.2 calculate electrostatic potentials(ESP) for dimethylphosphate(DMP) and modified nucleoside
input files: DMP_ESP.gjf mG_ESP.gjf
output files: DMP_ESP.out mG_ESP.out
1.3 convert the Gaussian ESP data into the RESP format
command: espgen -i mG_ESP.out -o mG.esp
espgen -i DMP_ESP.out -o DMP.esp
cat DMP.esp mG.esp> m2G_esp1.esp
1.4 run the RESP fit
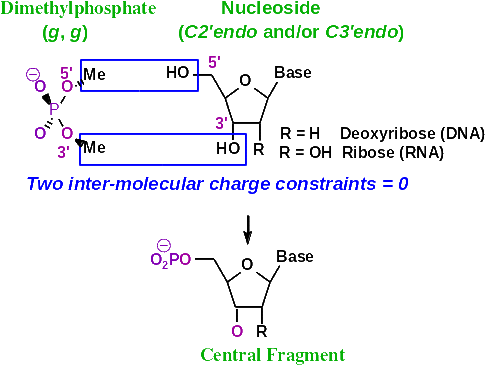
I set inter-molecular charge constraints between the two Dimethylphosphate methyl atoms and the HO3' and HO5' hydroxyl atoms of a nucleoside. I also set the equivalent of sugar atoms that a modified nucleoside to a normal nucleoside, and some other constraints.
command1:resp -O -i m2G_resp1.in -o m2G_resp1.out -p m2G_resp1.pch -t m2G_resp1.chg -q m2G_resp1.qin -e m2G_esp1.esp
command2:resp -O -i m2G_resp2.in -o m2G_resp2.out -p m2G_resp2.pch -t m2G_resp2.chg -q m2G_resp1.chg -e m2G_esp1.esp
2.Build a modified nucleotide Leap library file and frcmod file containing custom parameters.
2.1 get the structure of modified nucleotide from PDB
file: Pre_m2G.pdb
2.2 enter the atom types and charges one by one in xleap,based by analogy to the ff14SB force field
2.3 save new library file
file: new_M2G.pdb M2G.lib
2.4 create a frcmod file
file: M2G.frcmod
Thank you in advance,
Mao
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: picture1.png)
