Date: Fri, 22 Feb 2019 14:17:57 +0300
Hi,
I am trying to parameterize dihedral angels according to AMBER7 manual
Appendix C . I will apply always idivf=1. According to the manual even if I
apply idivf=1 I have to divide "potantial" by number of torsion angels to
obtain frcmod Kb value. I am not sure if I understood right.
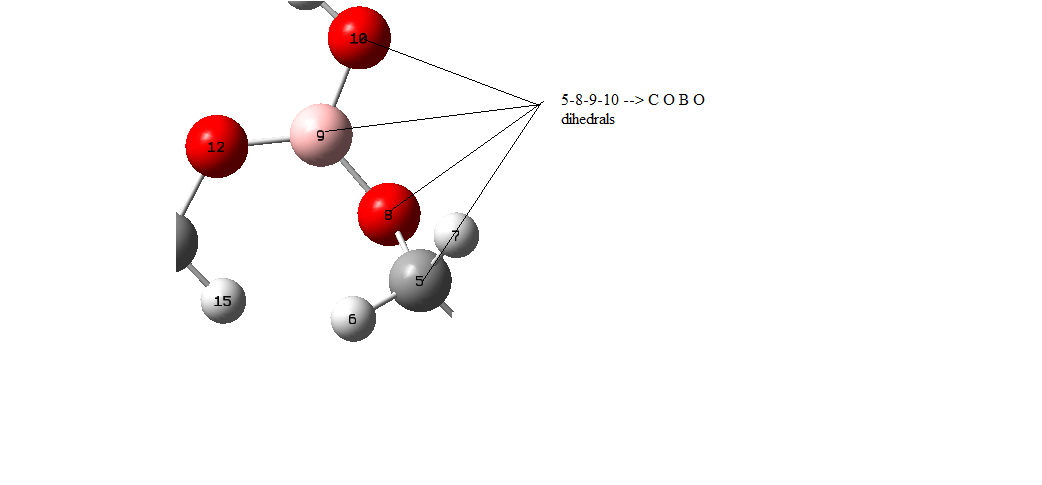
For examle in this molecule for this dihedral:
https://ibb.co/RzChG2N (image attached)
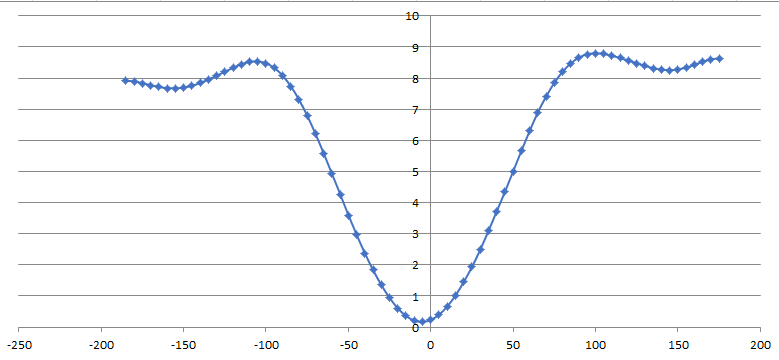
I scanned C-O-B-O dihedral and I obtained this chart:
https://ibb.co/pzFtqyt (image attached)
( in the chart: at 0 degree there is minimumum (teta=180 degree), PN=1 and
V=9 kcal/mol)
However in this molecule:
for C-O-B-O dihedral there are two sets ( 5-8-9-10 and 5-8-912 or B
attached two atoms) so if I understood true Kb= 9/2/2 = 2.25 kcal mol but
otherwise if I don't care this Kb=9/2=4.500 kcal/mol
here is the question should I put in frcmod file this value:
C -O - B - O 1 2.25 180 1 ( dihedral name - idivf - Kb - Teta -
PN) ...(1)
or this value:
C -O - B - O 1 4.50 180 1 ( dihedral name - idivf - Kb - Teta -
PN) ...(2)
I think (1) is true but I cound't be sure. Could you help me if I am wrong?
Thank you so much by now
.
NOTE: I attached images if you don't want to click links.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: cobo.png)
(image/png attachment: cobo_chart.png)
