Date: Mon, 4 Feb 2019 11:46:21 +0100
Hi David,
>Why? The difference is just a constant (for any given system). In what
>way does a change in the zero of energy change your analysis?
I totally agree with you.
This is why I am even more surprised when I make the difference of the same
snapshots total energy for the two different approcahes.
In principle I should get a constant for every point.
Instead I get large differences.
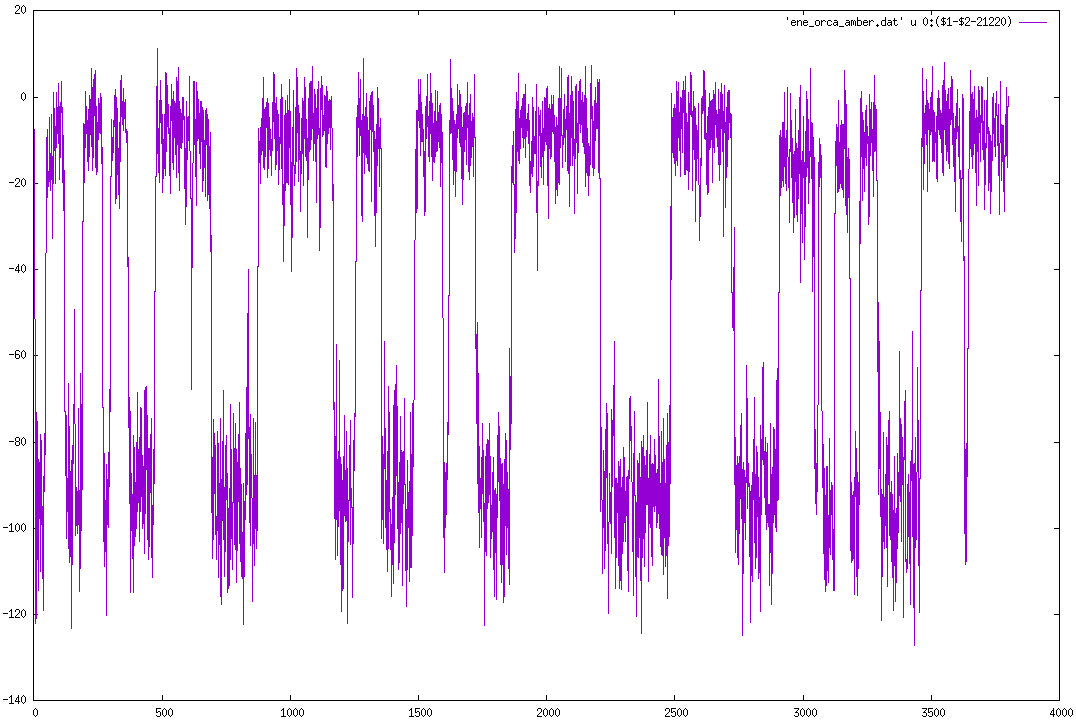
In the figure below I report this energy difference for a reactive
trajectory of the SN2 reaction CH3Cl +F- -> CH3F + Cl-
[image: Screenshot from 2019-02-04 10-14-26.png]
I shifted the data of 21220 kcal/mol to make it more readable.
I am puzzled, maybe I am making a terrible mistake but I do not really
understand where.
I attach the two directories with the inputs that I used to run the code.
If you have time to have a look it would be great.
I run the trajectory using the following sander command
mpirun sander.MPI -O -i 03_Prod.in -o 03_Prod.out -p solvated.prmtop -c
02_Heat.rst -y test_traj_stride.mdcrd -x 03_Prod.mdcrd -inf 03_Prod.info
therefore it should be straightforward to test.
I hope this is not too annoying for you.
Best,
Giovanni
amber_pm3.targz
<https://drive.google.com/file/d/1_fuSNDGS3JilHTQBZwNUurCbEs4fWXOb/view?usp=drive_web>
orca_pm3.targz
<https://drive.google.com/file/d/1MrF0UnzLTw7fbjsDms2kvTcuCzoOyvHz/view?usp=drive_web>
Il giorno lun 4 feb 2019 alle ore 02:22 David Case <david.case.rutgers.edu>
ha scritto:
> On Sun, Feb 03, 2019, GiovanniMaria Piccini wrote:
>
> >However, I really need the total potential energy like to one that I get
> >from the Orca PM3 output and not the heats of formations.
>
> Why? The difference is just a constant (for any given system). In what
> way does a change in the zero of energy change your analysis?
>
> >1) If one uses imin=5 reading an external trajectory with plumed=1 and a
> >plumed file, AMBER recognizes the presence of plumed and say it will use
> >it.
> >however, plumed is not called during the AMBER run with the external
> >trajectory and , thus, I had to grep the information I needed from the
> >AMBER output.
>
> This sounds like something we should look into. I'm guessing relatively
> few people actually use plumed with Amber (especially with imin=5), so I
> don't recall having seen this behavior reported before.
>
> >
> >2) If AMBER prints only the heats of formation than there is a problem
> with
> >the plumed2 patch as plumed intends to read the potential energy and
> >instead it read the heats of formation.
> >From the plumed side we can modify the patch ubt we nedd to have access to
> >the potential energy from AMBER.
>
> Same question as above here.
>
> ....dac
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_from_2019-02-04_10-14-26.png)
