Date: Mon, 19 Nov 2018 22:52:15 +0530
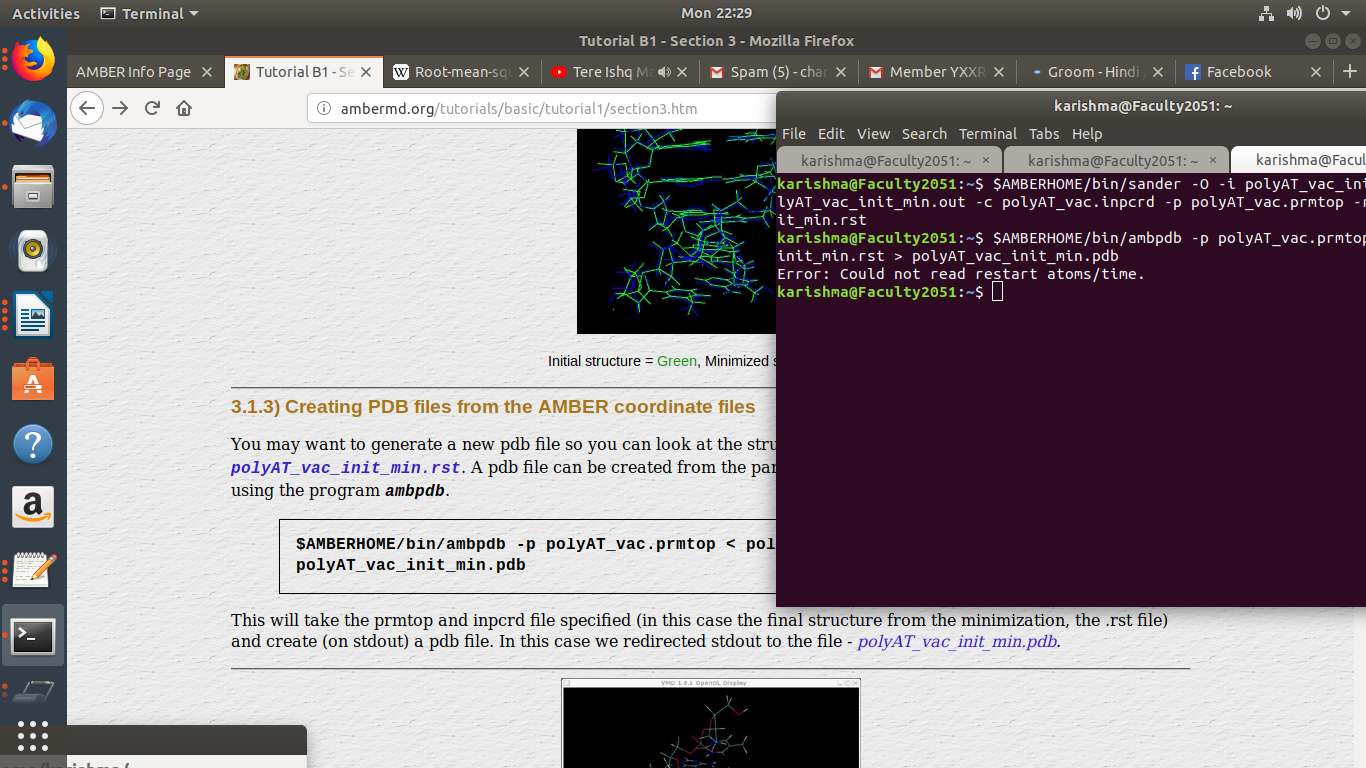
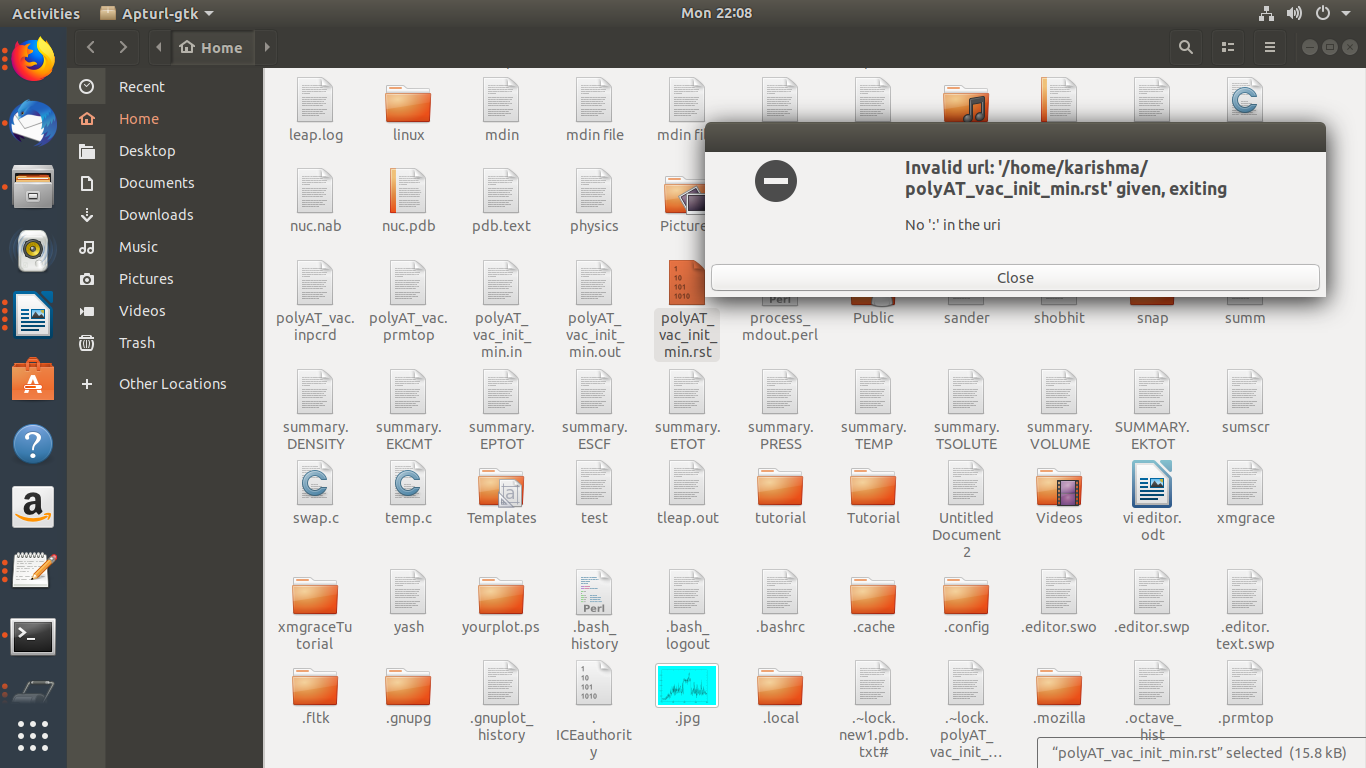
SIR WHY MY AMBPDB FILE NOT CREATING ? WHY ONLY MY polyAT_vac_init_min.rst
<http://ambermd.org/tutorials/basic/tutorial1/files/output_files/polyAT_vac_init_min.rst>.
SHOWING ERROR ?
On Mon, Nov 19, 2018 at 7:32 PM Daniel Roe <daniel.r.roe.gmail.com> wrote:
> Hi,
>
> Your issue is here:
>
> On Mon, Nov 19, 2018 at 7:59 AM Rajbinder Kaur Virk
> <rajbinderkaurvirk.gmail.com> wrote:
> > No clusters found.
>
> This isn't a cpptraj error per se. Based on the parameters you gave
> the 'cluster' command, no clusters were found. You'll need to try to
> adjust the parameters you're giving the DBSCAN algorithm, or try
> another algorithm. Clustering is much more of an art form than a
> science, so plan on a fair amount of trial and error here. It's good
> that you're starting off with a relatively small trajectory; this will
> allow you to become familiar with the results without waiting around
> too much. I highly recommend reading the entire 'cluster' command
> entry in the Amber 18 manual if you haven't already (section 29.11.4,
> starting on page 694), and in particular the 'Hints for setting DBSCAN
> parameters' subsection if you want to continue using DBSCAN. I also
> recommend reading this excellent paper on clustering MD trajectories
> from Shao, Cheatham et al.:
> https://pubs.acs.org/doi/abs/10.1021/ct700119m
>
> Some general clustering tips:
>
> 1) In my experience I've obtained better results with 'sieve <#>
> random' as opposed to plain 'sieve <#>'.
> 2) If you're not going to change your distance metric (including
> sieve) you way want to use 'savepairdist'/'loadpairdist' to avoid
> recalculating the pairwise distances every time. This is useful e.g.
> when doing hierarchical aggolmerative with varying epsilon, etc.
> 3) The OpenMP version of cpptraj (cpptraj.OMP) can accelerate the
> pairwise distance calculation, but don't use more threads than you
> have physical cores.
>
> Also, there is a very basic clustering tutorial you may find useful
> here: http://www.amber.utah.edu/AMBER-workshop/London-2015/Cluster/
>
> Hope this helps,
>
> -Dan
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_from_2018-11-19_22-29-26.png)
(image/png attachment: Screenshot_from_2018-11-19_22-08-30.png)
