Date: Sun, 18 Nov 2018 15:09:51 +0100
Hi all,
By reading these two interesting articles, I am wondering if similar
analysis are manageable using Amber tools as well..
https://www.ncbi.nlm.nih.gov/pubmed/28319010
https://www.ncbi.nlm.nih.gov/pubmed/27019343
We are interested for example in investigate through time evolution of the
MD the electrostatic, “hydrophobic” interaction and number of contacts
between a series of protein-protein molecular object..
And do you believe is more “efficient” dealing with PBC “issues” directly
with GROMACs or even the PBC re-imaging should be fixed with Ambertools?
Thank you for your possible suggestions!
[image: analysis.PNG]
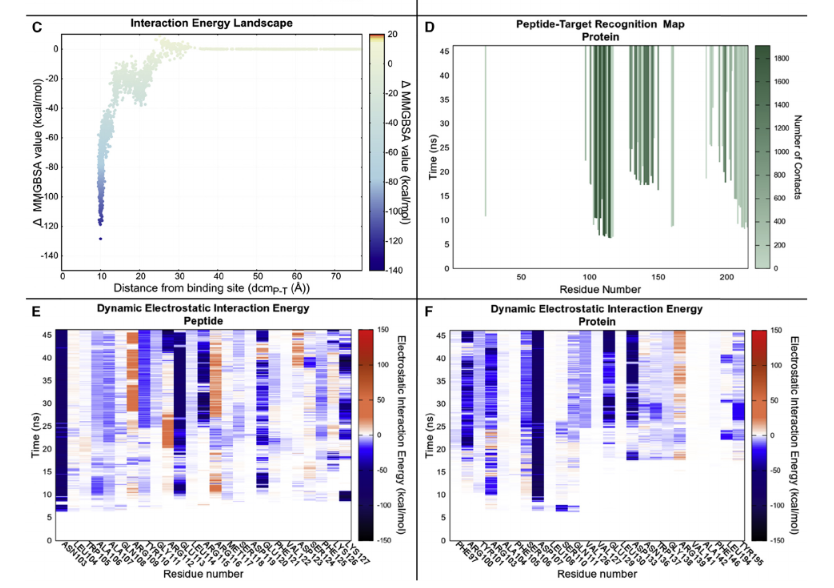
Figure. (D) Peptide-target recognition map.(E and F) Dynamic electrostatic
interaction energy, on the peptide and protein side, respectively.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: analysis.PNG)
