Date: Mon, 27 Aug 2018 11:46:36 +0530
Dear Amber Users,
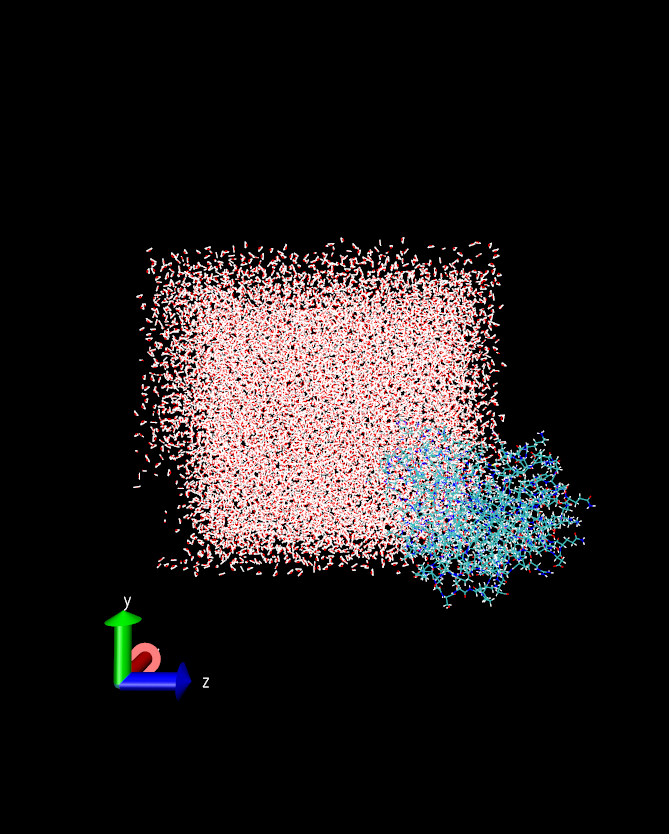
I am simulating a protein molecule containing
259 amino acid residues. I have followed the standard protocol of
simulation, minimization, heating, equilibration (in NVT and NPT) and then
production run (NVT) using a rectilinear simulation box and PMEMD.cuda.MPI.
Starting from the heating step I have been using the 'iwrap = 1' . After
few nanosecond of production run I have observed that a part of the protein
molecule has come out of the box. I have attached here an image file for
the understanding of the condition.
So my concern is if there is any possibility of altering the dynamics or
any other properties due to this situation. Should I put the protein
molecule back in the center of box whenever it comes out of the box? Or,
may I ignore this and continue the production run?
Any fruitful suggestion will be very helpful to me.
Thanking you.
Sanjib Paul
Research scholar
Department of Chemistry
Indian Institute of Technology Kharagpur
Kharagpur 721302
India
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: snap.jpeg)
