Date: Wed, 1 Aug 2018 13:45:46 -0400
Dear AMBER community,
For the past two months I have been trying to perform a simple MD
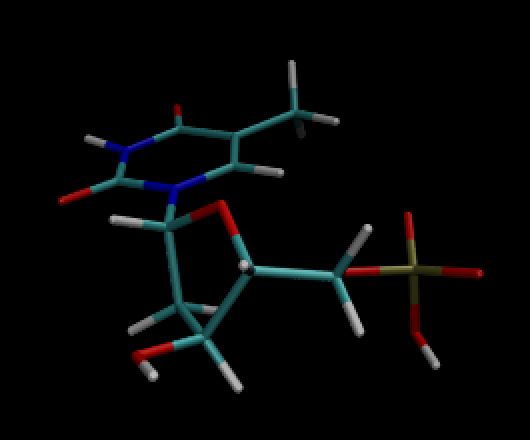
simulation on dTMP. Attached is a picture of the structure and the .inpcrd
and .prmtop files that I used. The issue that I am encountering is that my
structure is imploding upon the NPT heating and NPT equilibration
sections. It almost seems like I am encountering a parameter issue (the
bonds and angles are going crazy). The structure is totally fine prior to
the heating phase (after minimizing the solvent, hydrogens, etc)
The system is literally: 1 dTMP, 0.15 M Na/Cl, and a water box.
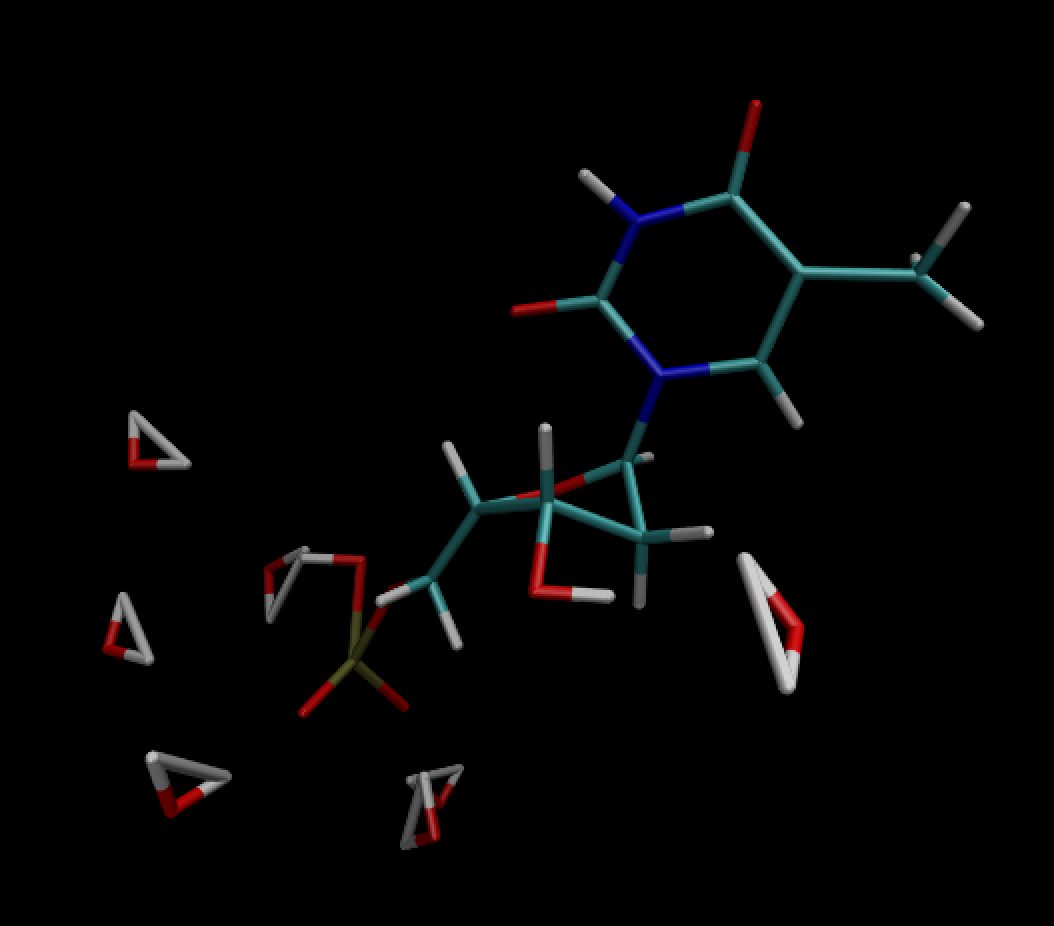
Prior to NPT heating the structure looks like this. Which looks fine.
The following is after NPT heating (all residues within 3 angstroms of
resid 1 (dTMP) is shown)
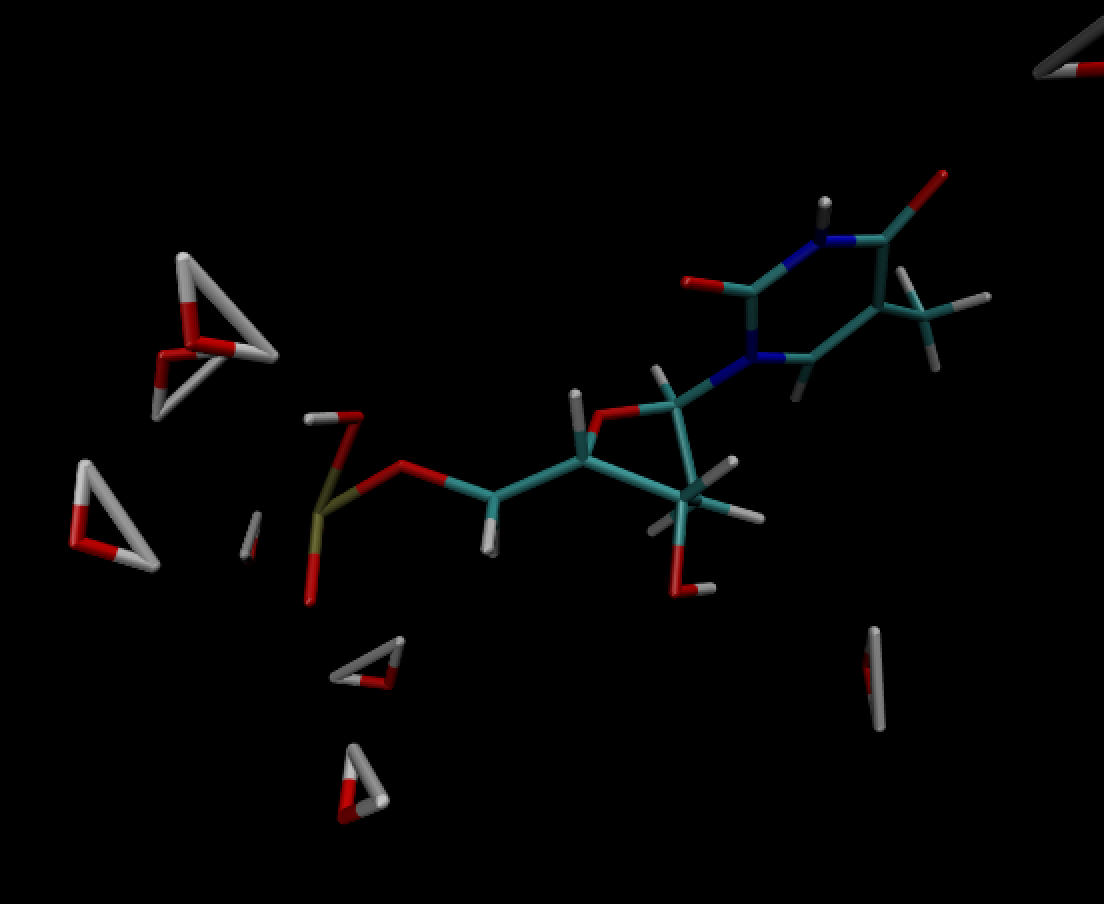
This is another angle to see the distortion better (same frame)
The following is after NPT equilibration.
The following is my npt_heat.in file:
First NPT running
&cntrl
imin=0, irest=0,
nstlim=360000, dt=0.001, ntx=1,
ntpr=500, ntwx=5000, ntwr=5000,
cut=10, ntb=2, ntp=1, pres0=1.01325,
ntc=2, ntf=2,
ntt=3, gamma_ln=2.0, ig=-1,
tempi=0.0, temp0=300.0,
ioutfm=1, ntwv=-1, ig=-1, iwrap=1,
nmropt=1,
/
&wt type='TEMP0', istep1=0, istep2=10000, value1=0.0, value2=50.0 /
&wt type='TEMP0', istep1=10001, istep2=60000, value1=50.0, value2=50.0 /
&wt type='TEMP0', istep1=60001, istep2=70000, value1=50.0, value2=100.0 /
&wt type='TEMP0', istep1=70001, istep2=120000, value1=100.0, value2=100.0 /
&wt type='TEMP0', istep1=120001, istep2=130000, value1=100.0, value2=150.0 /
&wt type='TEMP0', istep1=130001, istep2=180000, value1=150.0, value2=150.0 /
&wt type='TEMP0', istep1=180001, istep2=190000, value1=150.0, value2=200.0 /
&wt type='TEMP0', istep1=190001, istep2=240000, value1=200.0, value2=200.0 /
&wt type='TEMP0', istep1=240001, istep2=250000, value1=200.0, value2=250.0 /
&wt type='TEMP0', istep1=250001, istep2=300000, value1=250.0, value2=250.0 /
&wt type='TEMP0', istep1=300001, istep2=310000, value1=250.0, value2=300.0 /
&wt type='TEMP0', istep1=310001, istep2=360000, value1=300.0, value2=300.0 /
&wt type='END' /
The following is my npt_eq.in file:
NPT equilibrium 2ns
&cntrl
imin=0, irest=1,
nstlim=2000000, dt=0.001, ntx=7,
ntpr=1000, ntwx=1000, ntwr=1000,
cut=10, ntb=2, ntp=1, pres0=1.01325,
ntc=2, ntf=2,
ntt=3, gamma_ln=1.0,
tempi=300.0, temp0=300.0,
ioutfm=1, ntwv=-1, ig=-1, iwrap=1,
I have tried recreating my .inpcrd and .prmtop files and still encounter
the same issue. I usually run using AMBER CUDA, so I tried running this
with CPUs, but that didn't work either. I am using Amber14/Ambertools
15.
Thank you for your help,
David
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber

(image/png attachment: Screenshot_2018-07-25_11.33.44.png)
(image/png attachment: Screenshot_2018-07-25_11.31.55.png)
(image/png attachment: Screenshot_2018-07-25_11.32.20.png)
(image/png attachment: Screenshot_2018-07-25_11.40.15.png)
