Date: Mon, 9 Jul 2018 20:37:11 +0200
Dear Amber users,
While carrying out an aMD simulation in chloroform solvent of my peptide,
only dihedral boost is applied even after iamd=3 (dual boost option).
The Ethresh value when lower, the calculation goes without any error except
that the boost energy is 0 kcal/mol at every step.
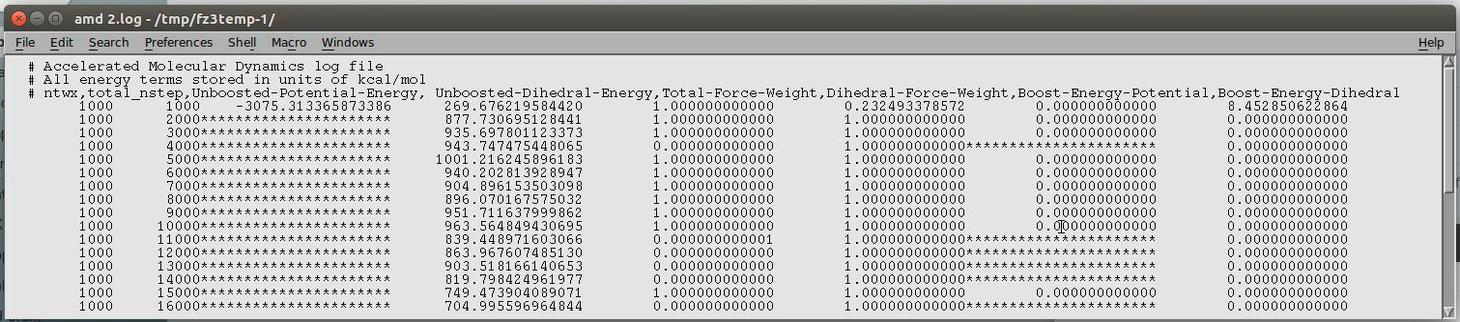
When the Ethresh value is higher than the average potential energy (as it
should be), the calculation proceeds but the output amd.log file looks like
this.
[image: image.png]
Can anyone explain what's wrong here? Ideally, for the boost to be applied
to the system, potential energy should be below the specified criterion, in
this case the Ethresh. Why should it show errors?
Any help would be appreciated.
-- Best wishes Chetna
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
