Date: Wed, 16 May 2018 17:10:19 +0200
Good afternoon
I am an organic chemist, with a little experience of comp chem . Till now I
worked with gaussian software and DFT model chem to compute activation
barriers ( work published in Catalysis Science & Technology 7 (7), 1497-1507
, <javascript:void(0)> Substituent and catalyst effects on GAC
lactonization of γ-hydroxy esters)
However, recently I computed strain energies from reactant to transition
state by using the DIASM model of Hook and Bickelhaupt. Since I would like
to rationalize these strain energies in terms of stretching , bending and
torsion, for this purpose I need to know the key parameters, i.e. force
constants. I decided to use AMBER, in order to evaluate these parameters ,
having read the interesting tutorial
"Using Antechamber to Create LEaP Input Files for Simulating Sustiva
(efavirenz)-RT complex using the General Amber Force Field" by Ross Walker
and Sishi Tang.
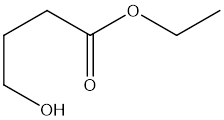
My idea is to input my DFT-optimized structure, say of a reactant, like
4-hydroxybutanoate
to Antechamber and Leap and get the pmrtop file with the set of GAFF
parameters which I need.
Do You think that this procedure is reasonable and feasible? If Yes , how
can be implemented in a batch of AMBER commands? I am a beginner of AMBER
and so I need the help of an expert.
Giuseppe Gatti , University Urbino
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- application/octet-stream attachment: oledata.mso
(image/png attachment: image003.png)
