Date: Fri, 23 Mar 2018 14:16:30 -0300
In the tutorial (3.1.3 creating PDB files from the AMBER coordinate
files), the following command should be used:
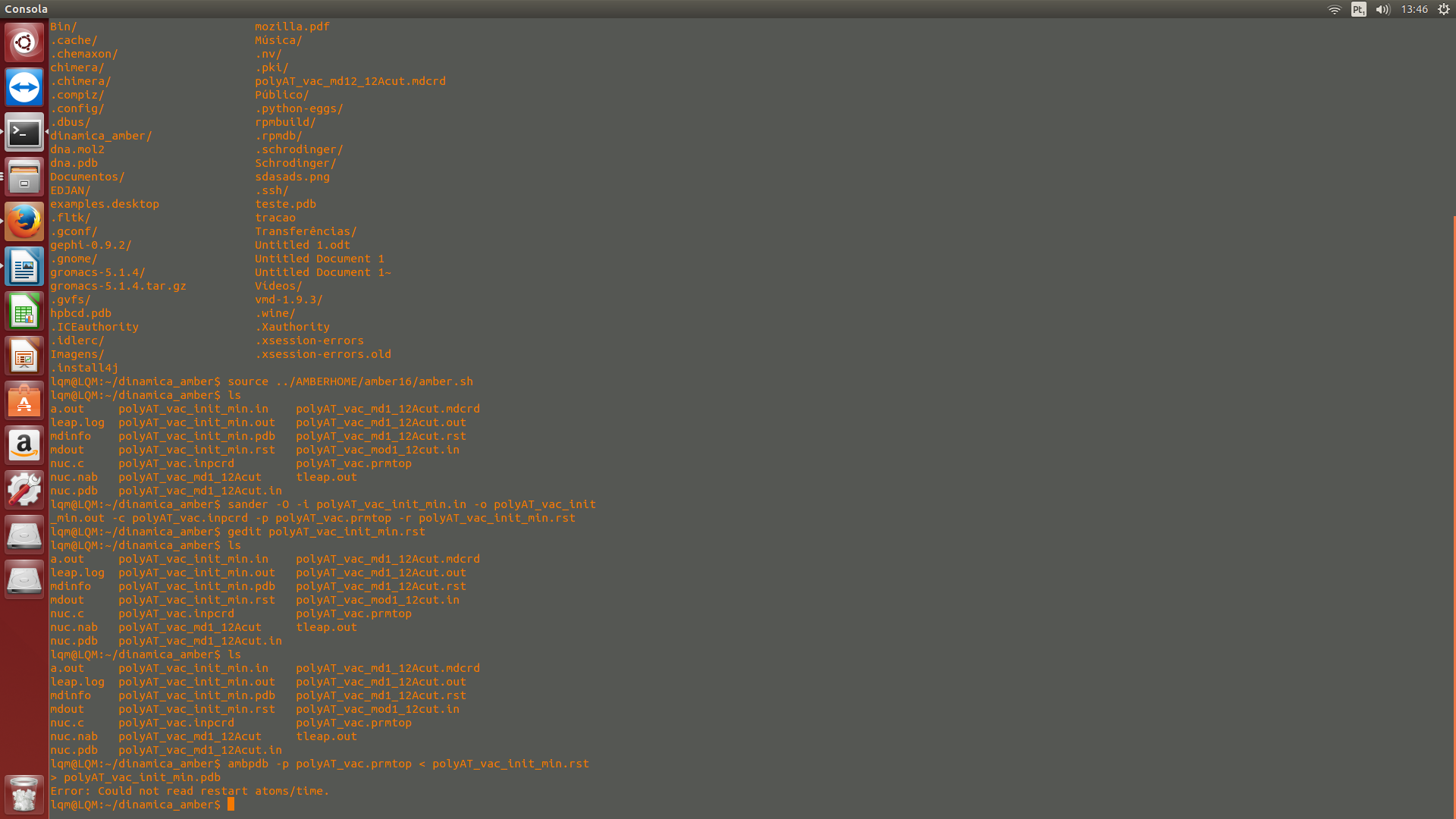
$ AMBERHOME / bin / ambpdb -p polyAT_vac.prmtop
<polyAT_vac_init_min.rst> polyAT_vac_init_min.pdb
After using this command the following message appears:
"Error: Could not read restart atoms / time."
I've done all the steps correctly. in an attachment, input files.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- application/octet-stream attachment: polyAT_vac.prmtop
- application/octet-stream attachment: polyAT_vac_md1_12Acut.rst
(image/png attachment: Screenshot_from_2018-03-23_13:46:41.png)
