Date: Fri, 1 Dec 2017 12:29:41 -0500
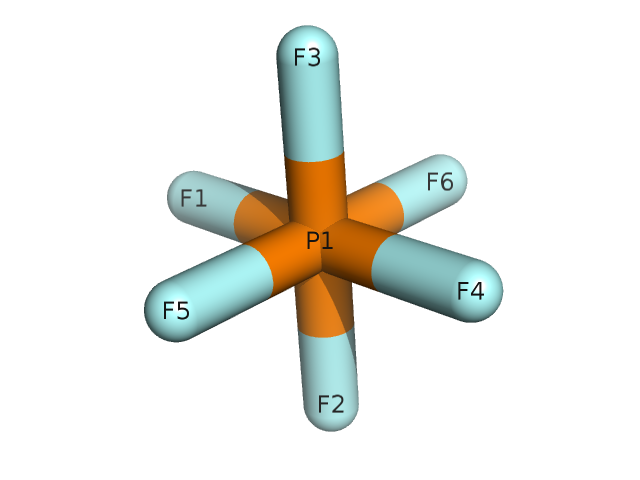
Here is my solution to discriminate the two types of f-p-f angles. See the
attached picture,
f1-p-f1, f2-p-f2 and f3-p-f3 have a reference bond angle of 180 and others
(f1-p-f2, f1-p-f3 and f2-p-f3) have a reference bond angle of 90.
You can easily add f1, f2 and f3 to $AMBERHOME/dat/antechamber/PARMCHK.DAT
(see attahcment) to make parmchk2 work.
I have also attached the prepi and frcmod files for this molecule.
All the best
Junmei
On Thu, Nov 30, 2017 at 7:00 PM, Sanaa ALAbbad <salabbad79.gmail.com> wrote:
> Dear Ambers,
>
> I have an issue with PF6- conformational structure. During the
> equilibration step, the octahedral geometry destroyed to square pyramidal.
> I first used the frcmod generated by AMBER
>
> remark goes here
> MASS
> p5 30.970 1.538
> f 19.000 0.320
>
> BOND
> p5-f 248.60 1.646
>
> ANGLE
> f -p5-f 44.040 90.000
>
> DIHE
>
> IMPROPER
>
> NONBON
> f 1.7460 0.0610
> p5 2.0940 0.2000
>
> and then I included the 180 angle and use the parameters used by Phys.
> Chem. Chem. Phys., 2003,5, 3481-3488
> that destroyed the structure even more
>
> remark goes here
> MASS
> p5 30.970 1.538
> f 19.000 0.320
>
> BOND
> p5-f 260.30 1.646
>
> ANGLE
> f -p5-f 194.100 90.000
> f -P5-f 194.100 180.00
>
> DIHE
>
> IMPROPER
>
> NONBON
> f 1.7460 0.0610
> p5 2.0940 0.2000
>
> Yet no luck!!
>
> I tried to use PARAMFIT but it defined only one angle of 90.
>
> Could any one help me with this regard?
>
> Thanks in advance.
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: PF6.png)
- application/octet-stream attachment: PARMCHK.DAT
- application/octet-stream attachment: PF6.prepi
- application/octet-stream attachment: PF6.prmtop
- application/octet-stream attachment: PF6.prmcrd
- application/octet-stream attachment: PF6.frcmod
