Date: Fri, 24 Nov 2017 11:18:32 +0530
Dear Sir/Ma'am
When i have made the topology and coordinate file, it is okay.
But the geometry of Glucose at O5-OH is in equilibration, not in
minimization.
For preparing frcmod file, I first used the conversion of Gaussian output
into the mol2 file then by the help of parmchk command made frcmod file.
I have extracted the library of 0GA+ROH from GLYCAM_06j-1. i prepared the
.lib file from following step.
tleap -s -f leaprc.GLYCAM_06j-1
GLC = sequence { ROH 0GA }
check GLA
saveamberparm GLA GLA.parmtop GLA.inpcrd
saveoff GLA GLA.lib
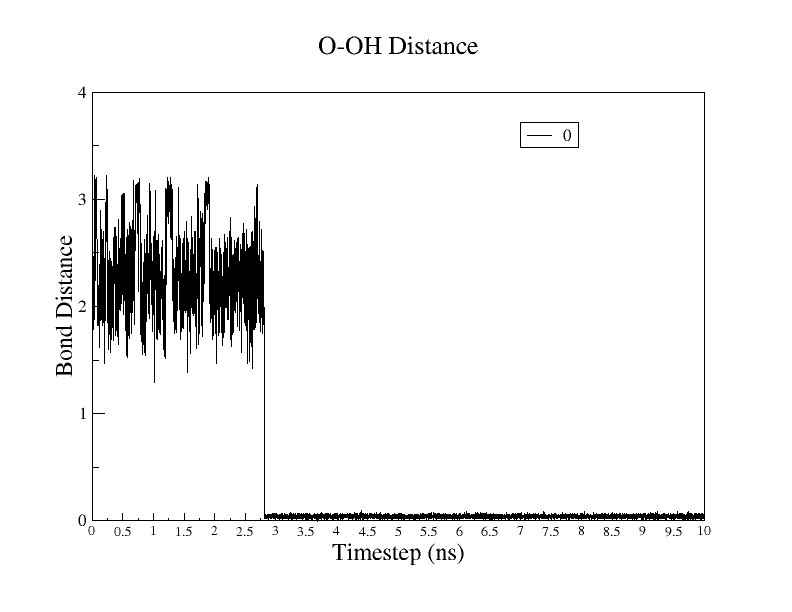
In the 0GA+ROH library,OH-O is not binding in the preparation of topology
and coordinate file. but is getting bound in equilibration.
I have also attached the binding graph of OH-O.
Regards
Aashish
On Wed, Nov 22, 2017 at 8:42 PM, Lachele Foley <lf.list.gmail.com> wrote:
> So far, you have explained how you calculated charges. Charge calculations
> should not alter the 3D structure, so the procedure you outline would not
> produce that geometry. At what specific step did the strange orientation
> occur? Did you minimize the structure? If so, was it there before or
> after minimization? If not, then what did you do? We need enough detail
> for this to repeat exactly what you did, and this is not enough.
>
> You say:
> "i have created glucose structure and optimized the same in Gaussian with
> b3lyp/6-31* method"
>
> You created the entire 3D structure of the glucose there? That calculation
> should not use any of the force field parameters. What was the structure
> at the end of that calculation? Was the hydrogen already in that
> orientation?
>
> You say:
> "I used simple glucose molecule because the parameter of glucose is already
> available in amber."
>
> Did you use the glycam06 parameters?
>
>
> On Tue, Nov 21, 2017 at 11:48 PM, Aashish Bhatt <
> aashish.ph16221.inst.ac.in>
> wrote:
>
> > Dear Sir/Ma'am
> >
> > I need to specify that i have executed this calculation for better
> > understanding of force field. So I used simple glucose molecule because
> the
> > parameter of glucose is already available in amber.
> > In the next step, i have created glucose structure and optimized the same
> > in Gaussian with b3lyp/6-31* method. After optimization, i have
> calculated
> > the ESP charge from #P HF/6-31G* SCF=Tight Pop=MK
> > IOp(6/33=2,6/41=10,6/42=17) method. I have used resp charge by the
> > "*antechamber
> > -i quer.log -fi gout -o quer.mol2 -fo mol2 -c resp" *command. Afterwards
> i
> > prepared the .lib file and then made topology and coordinate file.
> >
> > Regards
> > Aashish
> >
> >
> > On Tue, Nov 21, 2017 at 7:26 PM, Lachele Foley <lf.list.gmail.com>
> wrote:
> >
> > > You will definitely get different charges if you do different types of
> > > calculations to obtain them. There are no 'correct' charges. In
> > physical
> > > actuality, partial charges do not exist at all. The ones that were
> > > developed for GLYCAM06 were developed to work with the rest of the
> force
> > > field. If you change those charges, it's hard to say what sort of
> > results
> > > you will get. If you get bad results, then either your charges are not
> > > good or there is some other problem with the way you're setting up the
> > > system.
> > >
> > > Why are you making a lib file for GLC? You can make a topology and
> > inpcrd
> > > file for glucose using the method described in the earlier thread. Why
> > not
> > > just use that method? Are you doing that just so you can change the
> > > charges? If so, then just make alternate versions of the prep files
> with
> > > your different charges. Doing that eliminates one source of possible
> > > error.
> > >
> > > If the anomeric OH binds to the O5, then there is something wrong with
> > the
> > > set of parameters you have constructed. Why not use the ones that
> > already
> > > exist? What do you want to do that makes you want to use Gaussian and
> > > calculate new charges?
> > >
> > > In any case, if you observe unusual behavior, we need to know exactly
> > what
> > > you did before we can help. We need a lot more information than what
> you
> > > have given us.
> > >
> > >
> > > On Mon, Nov 20, 2017 at 7:00 AM, Aashish Bhatt <
> > aashish.ph16221.inst.ac.in
> > > >
> > > wrote:
> > >
> > > > Dear Sir/ Ma'am
> > > >
> > > > I am facing a problem in 0GA+ROH carbohydrate parameter while doing
> > force
> > > > field preparation for glucose molecule. I have used Gaussian
> optimized
> > > > structure and calculated the MK charges(Gaussian) and resp
> > > > charges(antechamber). Then I compared the results with 0GA+ROH.dat
> file
> > > and
> > > > found differences between the two. I have created the library file
> > > resp.lib
> > > > and mk.lib for preparation of topology and coordinate file for
> Glucose
> > > > molecule. I observed that the anomeric OH binds to the O5 atom of
> > Glucose
> > > > in both vacuum and solvated phase. I don't know the reason behind
> this
> > > > unusual binding behavior. Kindly help me if there is any charge
> related
> > > > issue of atoms in library file.
> > > >
> > > > I am attaching both the library files along with 0GA+ROH.dat.
> > > >
> > > > Thanking You
> > > > In wait of your kind reply.
> > > >
> > > > With regards
> > > >
> > > > Aashish
> > > >
> > > > _______________________________________________
> > > > AMBER mailing list
> > > > AMBER.ambermd.org
> > > > http://lists.ambermd.org/mailman/listinfo/amber
> > > >
> > > >
> > >
> > >
> > > --
> > > :-) Lachele
> > > Lachele Foley
> > > CCRC/UGA
> > > Athens, GA USA
> > > _______________________________________________
> > > AMBER mailing list
> > > AMBER.ambermd.org
> > > http://lists.ambermd.org/mailman/listinfo/amber
> > >
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
>
>
>
> --
> :-) Lachele
> Lachele Foley
> CCRC/UGA
> Athens, GA USA
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: O-OH_bond.png)
