Date: Mon, 16 Oct 2017 16:00:30 -0700
This question is regarding the R.E.D. server and has been posted on the
q4md mailing list, but I am also posting it here to solicit expert advice.
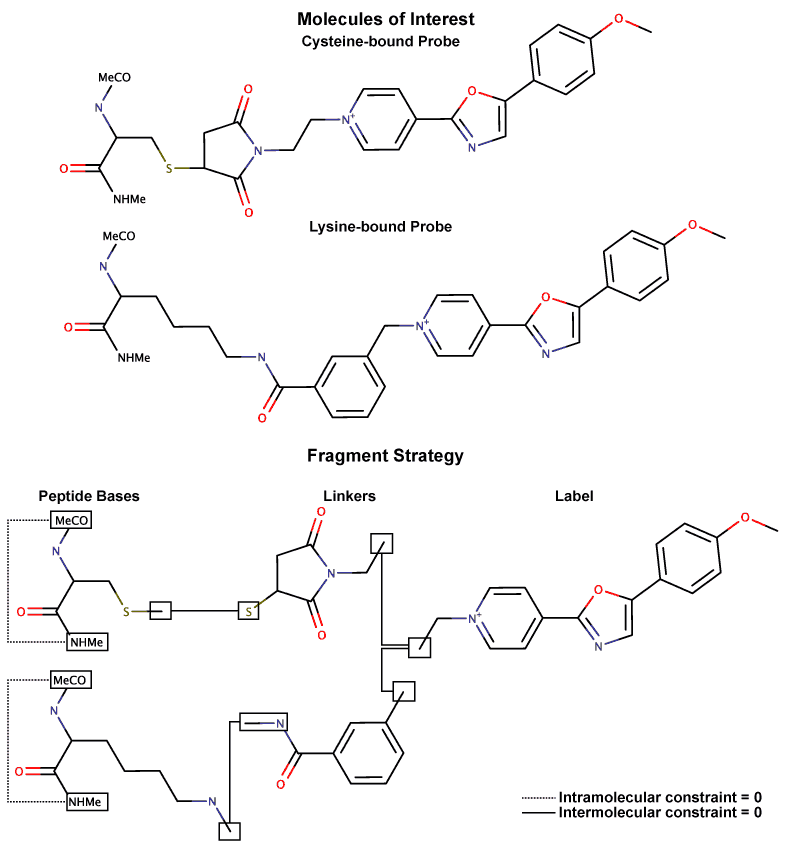
I am endeavoring to derive RESP charges for a fluorescent probe/dye
covalently bound to either a cysteine or a lysine, each with its own linker
(see attached schema.gif). After reading through the R.E.D. tutorial and a
number of R.E.DD.B. projects, I have developed the following proposed
approach (see attached schema):
Using a fragment-based approach, I divide each target molecule into three
fragments (dipeptide, linker, probe), where the probe is common for both
molecules. Rather than take existing Cys and Lys charges, I re-fit them
using a dipeptide and capping groups. Suitable capping groups are selected
for each fragment, and intra- and inter-molecular charge constraints are
used. Due to the inherent symmetry in the probe and its preference for
planarity, I consider it as a single fragment with charge +1 and only one
conformer.
How do this fragment approach and the corresponding capping groups look?
Should the probe be separated into smaller fragments? To generate the
entire molecule, should each assembled molecule of interest
(dipeptide-linker-probe) also be included in the optimization? Lastly, it
is my understanding that R.E.D. does not derive missing angle and torsion
parameters but relies on the user to provide them by analogy. Is
antechamber the best approach for obtaining these analogies if none can be
found in the literature?
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/gif attachment: scheme.gif)
