Date: Mon, 25 Sep 2017 09:47:51 +0000
Hi All,
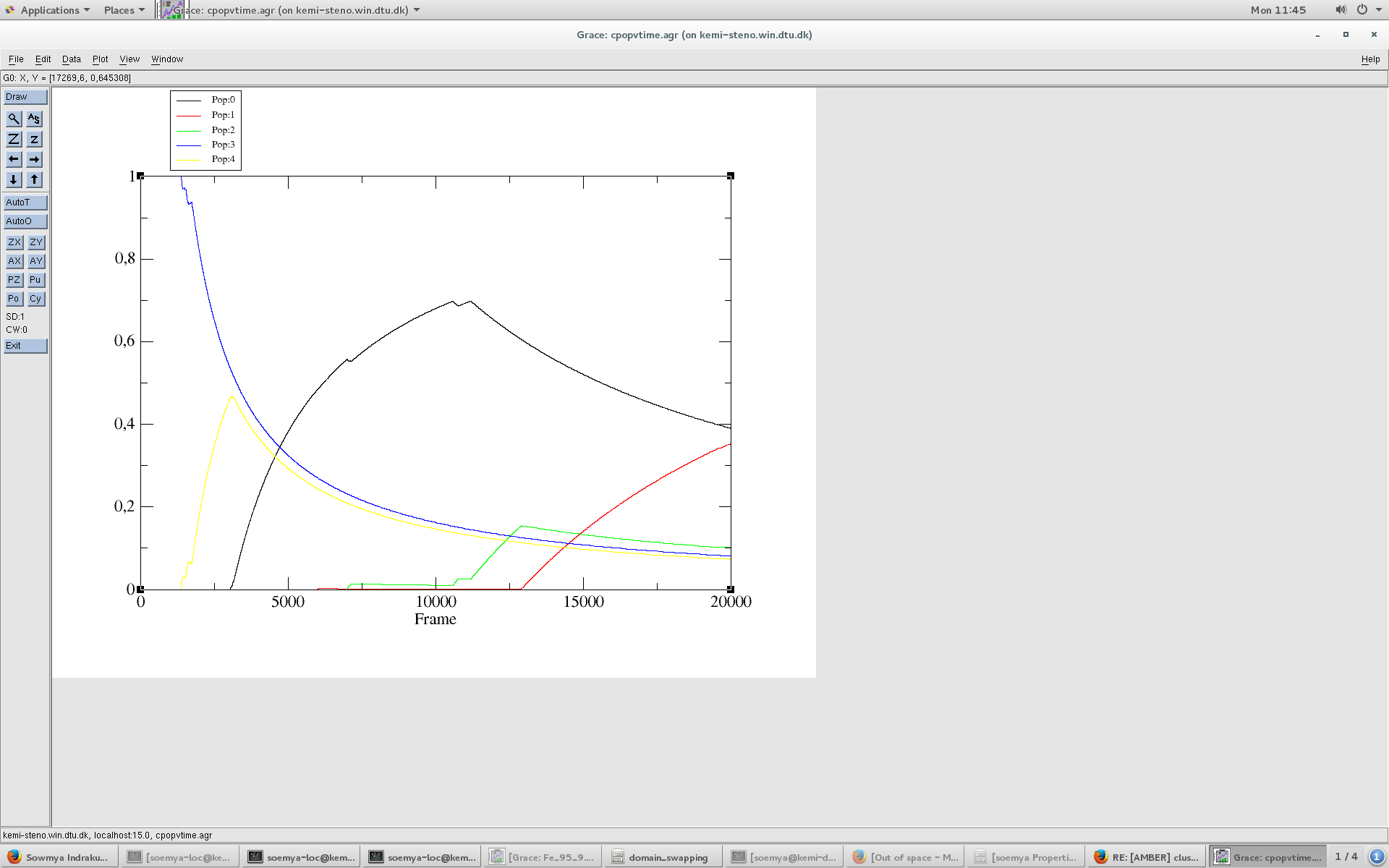
This works fine with the average linkage algorithm. I see a wide distribution of structures which is also observed in the RMSD and RMSF. (please see cluster population VS time plot attached).
I would really like to know why dbscan did not work.
Thanks
Regards
Sowmya
________________________________
From: Sowmya Indrakumar
Sent: Monday, September 25, 2017 11:38 AM
To: AMBER Mailing List
Subject: RE: [AMBER] cluster analysis using cpptraj
Hi Bill,
This is the script I'm running.
trajin ../../5.0/prod_pH.nc
strip :WAT,Cl-,Na+ outprefix noions
cluster C0 \
dbscan minpoints 10 epsilon 0.5 sievetoframe \
rms :1-677.CA,N,C \
sieve 100 \
out cnumvtime.dat \
summary summary.dat \
info info.dat \
cpopvtime cpopvtime.agr normframe \
repout rep repfmt pdb \
singlerepout singlerep.nc singlerepfmt netcdf \
avgout Avg avgfmt restart
I too suspect that it cannot find a cluster. I am trying other clustering method (average linkage rms based also).
Regards
Sowmya
________________________________________
From: Bill Ross [ross.cgl.ucsf.edu]
Sent: Monday, September 25, 2017 11:30 AM
To: amber.ambermd.org
Subject: Re: [AMBER] cluster analysis using cpptraj
> No clusters found.
That may be more relevant, in which case beyond me.
Bill
On 9/25/17 2:28 AM, Bill Ross wrote:
> Going on first principles with no knowledge of cpptraj, did you define
> any output files in your .in file?
>
> Bill
>
>
> On 9/25/17 2:22 AM, Sowmya Indrakumar wrote:
>> Dear All,
>> I am doing cluster analysis using the following script from: http://www.amber.utah.edu/AMBER-workshop/London-2015/Cluster/
>>
>>
>> I get this message after it's complete with no output file. I even changed the cutoff of minpoints and epsilon, it still gives the same message.
>>
>> cpptraj ../../model_sol.mod.parm7 cpptraj.in
>>
>> CPPTRAJ: Trajectory Analysis. V17.00
>> ___ ___ ___ ___
>> | \/ | \/ | \/ |
>> _|_/\_|_/\_|_/\_|_
>>
>> | Date/time: 09/25/17 10:55:25
>> | Available memory: 3.626 GB
>>
>> Reading '../../model_sol.mod.parm7' as Amber Topology
>> Radius Set: H(N)-modified Bondi radii (mbondi2)
>> INPUT: Reading input from 'cpptraj.in'
>> [trajin ../../5.0/prod_pH.nc]
>> Reading '../../5.0/prod_pH.nc' as Amber NetCDF
>> [strip :Na+ outprefix noions]
>> STRIP: Stripping atoms in mask [:Na+]
>> Stripped topology will be output with prefix 'noions'
>> [cluster C0 dbscan minpoints 25 epsilon 0.9 sievetoframe rms :1-677.CA,N,C sieve 100 out cnumvtime.dat summary summary.dat info info.dat cpopvtime cpopvtime.agr normframe repout rep repfmt pdb singlerepout singlerep.nc singlerepfmt netcdf avgout Avg avgfmt restart]
>> CLUSTER: Using coords dataset _DEFAULTCRD_, clustering using RMSD (mask [:1-677.CA,N,C]) best-fit
>> DBSCAN:
>> Minimum pts to form cluster= 25
>> Cluster distance criterion= 0.900
>> Sieved frames will only be added back if they are within
>> 0.900 of a frame in an existing cluster.
>> (This option is more accurate and will identify sieved
>> frames as noise but is slower.)
>> Initial clustering sieve value is 100 frames.
>> Only non-sieved frames will be used to calc within-cluster average.
>> Cluster # vs time will be written to cnumvtime.dat
>> Cluster pop vs time will be written to cpopvtime.agr (normalized by frame)
>> Pairwise distance data set is 'C0[PWD]'
>> Cluster information will be written to info.dat
>> Summary of cluster results will be written to summary.dat
>> Representative frames will be chosen by closest distance to cluster centroid.
>> Cluster representatives will be written to 1 traj (singlerep.nc), format Amber NetCDF
>> Cluster representatives will be written to separate trajectories,
>> prefix (rep), format PDB
>> Average structures for clusters will be written to Avg, format Amber Restart
>> Warning: One or more analyses requested creation of default COORDS DataSet.
>> CREATECRD: Saving coordinates from Top model_sol.mod.parm7 to "_DEFAULTCRD_"
>> ---------- RUN BEGIN -------------------------------------------------
>>
>> PARAMETER FILES (1 total):
>> 0: model_sol.mod.parm7, 82378 atoms, 24759 res, box: Orthogonal, 24083 mol, 23978 solvent
>>
>> INPUT TRAJECTORIES (1 total):
>> 0: 'prod_pH.nc' is a NetCDF AMBER trajectory, Parm model_sol.mod.parm7 (Orthogonal box) (reading 20000 of 20000)
>> Coordinate processing will occur on 20000 frames.
>>
>> BEGIN TRAJECTORY PROCESSING:
>> .....................................................
>> ACTION SETUP FOR PARM 'model_sol.mod.parm7' (2 actions):
>> 0: [strip :Na+ outprefix noions]
>> Stripping 45 atoms.
>> Stripped topology: 82333 atoms, 24714 res, box: Orthogonal, 24038 mol, 23978 solvent
>> Writing topology 0 (model_sol.mod.parm7) to 'noions.model_sol.mod.parm7' with format Amber Topology
>> 1: [createcrd _DEFAULTCRD_]
>> Warning: COORDS data sets do not store times.
>> Estimated memory usage (20000 frames): 19.760 GB
>> ----- prod_pH.nc (1-20000, 1) -----
>> 0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Complete.
>>
>> Read 20000 frames and processed 20000 frames.
>> TIME: Avg. throughput= 46.0936 frames / second.
>>
>> ACTION OUTPUT:
>>
>> ANALYSIS: Performing 1 analyses:
>> 0: [cluster C0 dbscan minpoints 25 epsilon 0.9 sievetoframe rms :1-677.CA,N,C sieve 100 out cnumvtime.dat summary summary.dat info info.dat cpopvtime cpopvtime.agr normframe repout rep repfmt pdb singlerepout singlerep.nc singlerepfmt netcdf avgout Avg avgfmt restart]
>> Starting clustering.
>> Mask [:1-677.CA,N,C] corresponds to 2031 atoms.
>> Estimated pair-wise matrix memory usage: > 79.664 kB
>> Pair-wise matrix set up with sieve, 20000 frames, 200 sieved frames.
>> Calculating pair-wise distances.
>> 0% 10% 20% 30% 40% 50% 60% 70% 80% 90% Complete.
>> Memory used by pair-wise matrix and other cluster data: 160.512 kB
>> 0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Complete.
>> No clusters found.
>> Cluster timing data:
>> TIME: Cluster Init. : 0.0011 s ( 0.01%)
>> TIME: Pairwise Calc.: 7.5530 s ( 73.76%)
>> TIME: Clustering : 0.0010 s ( 0.01%)
>> TIME: Cluster Post. : 0.0000 s ( 0.00%)
>> TIME: Cluster renumbering/sieve restore 0.0000 s ( 0.00%)
>> TIME: Find best rep. 0.0000 s ( 0.00%)
>> TIME: Info calc 0.0000 s ( 0.00%)
>> TIME: Summary calc 0.0000 s ( 0.00%)
>> TIME: Coordinate writes 0.0000 s ( 0.00%)
>> TIME: Total: 10.2406 s
>>
>> TIME: Analyses took 10.2480 seconds.
>>
>> DATASETS (3 total):
>> _DEFAULTCRD_ "_DEFAULTCRD_" (coordinates), size is 20000 (19.760 GB) Box Coords, 82333 atoms
>> C0 "C0" (integer), size is 0
>> C0[PWD] "C0[PWD]" (cluster matrix), size is 19900
>>
>> DATAFILES (2 total):
>> cnumvtime.dat (Standard Data File): C0
>> cpopvtime.agr (Grace File):
>> Warning: Set 'C0' contains no data.
>> Warning: File 'cnumvtime.dat' has no sets containing data.
>> Warning: File 'cpopvtime.agr' has no sets containing data.
>>
>> RUN TIMING:
>> TIME: Init : 0.0000 s ( 0.00%)
>> TIME: Trajectory Process : 433.8995 s ( 94.95%)
>> TIME: Action Post : 0.0424 s ( 0.01%)
>> TIME: Analysis : 10.2480 s ( 2.24%)
>> TIME: Data File Write : 0.0012 s ( 0.00%)
>> TIME: Other : 12.7649 s ( 0.03%)
>> TIME: Run Total 456.9560 s
>> ---------- RUN END ---------------------------------------------------
>> TIME: Total execution time: 457.5483 seconds.
>> --------------------------------------------------------------------------------
>> To cite CPPTRAJ use:
>> Daniel R. Roe and Thomas E. Cheatham, III, "PTRAJ and CPPTRAJ: Software for
>> Processing and Analysis of Molecular Dynamics Trajectory Data". J. Chem.
>> Theory Comput., 2013, 9 (7), pp 3084-3095.
>>
>>
>> Kindly, suggest what I'm doing wrong.
>>
>> Thanks
>> Regards
>> Sowmya
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_from_2017-09-25_11-45-00.png)
