Date: Mon, 11 Sep 2017 22:09:05 +0800 (GMT+08:00)
Hello everyone,
I read a paper in which the MD simulation of HP-beta-cyclodextrin was performed with Amber.
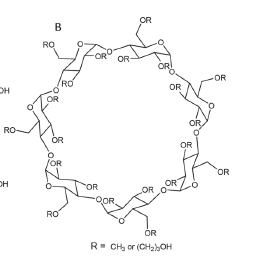
HP-beta-cyclodextrin is a cyclic oligosaccharide made up of seven dextrose units and seven hydroxypropyl groups (R group).
Here is the structure of HP-beta-cyclodextrin:
In this paper, the authors use GLYCAM_06j-1 for the dextrose units and GAFF force field for R groups separately. Since they are covalently connected with each other, can you tell me how to use different force fields for different parts of a molecule?
Any help from you will be greatly appreciated.
Yours sincerely,
Anhui
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: HP-beta-CD.png)
