Date: Sat, 26 Aug 2017 21:41:33 +0200
Dear AMBER users,
I need some advice about force field modification. I have a series of
ligands that have this imino-hydantoin moiety in common. The frcmod.ligand
file generated with parmchk has only IMPROPER angle definitions (is this
normal?).
remark goes here
> MASS
>
> BOND
>
> ANGLE
>
> DIHE
>
> IMPROPER
> c -c2-n -c3 1.1 180.0 2.0 Using default
> value
> n -n2-c2-nh 1.1 180.0 2.0 Using default
> value
> c2-c3-nh-hn 1.1 180.0 2.0 Using default
> value
> ca-ca-ca-ha 1.1 180.0 2.0 General
> improper torsional angle (2 general atom types)
> ca-cp-ca-ha 1.1 180.0 2.0 General
> improper torsional angle (2 general atom types)
> ca-ca-cp-cp 1.1 180.0 2.0 Using default
> value
> c3-n -c -o 10.5 180.0 2.0 General
> improper torsional angle (2 general atom types)
>
> NONBON
>
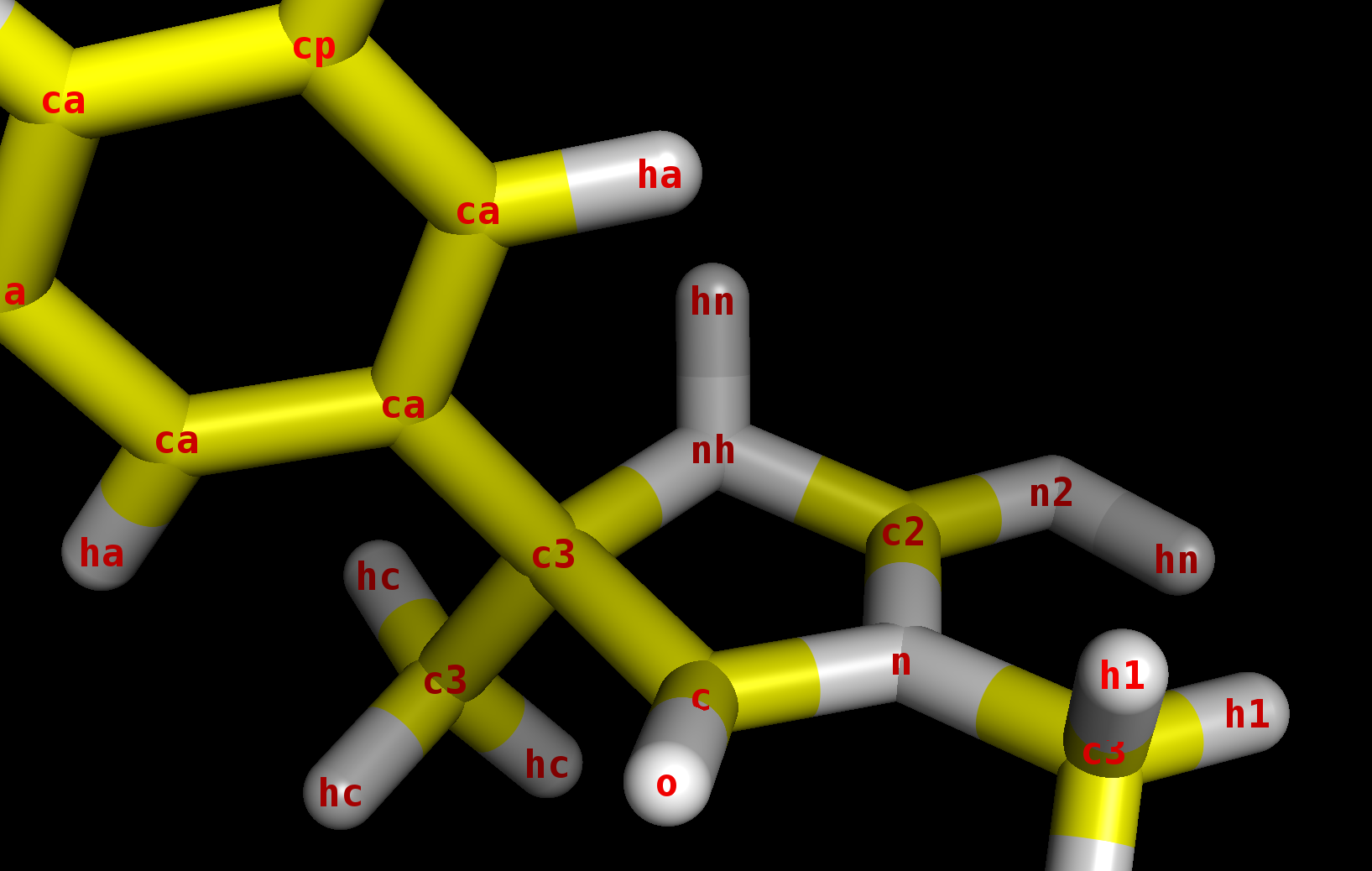
Particularly, the hn proton shown in the picture below is out of the place
of the aromatic imino-hydantoin ring. In the IMPROPER section, there is a
dihedral angle definition for that (c2-c3-nh-hn), therefore I wonder why it
does not remain on the aromatic plane during the MD simulation. What
modifications could I make in the frcmod file in order to maintain the
aromaticity of imino-hydantoin moiety?
https://www.dropbox.com/s/iblcyo6tz816x11/wrong_geometry.png?dl=0
Thanks in advance for any advice.
Thomas
--
======================================================================
Dr Thomas Evangelidis
Post-doctoral Researcher
CEITEC - Central European Institute of Technology
Masaryk University
Kamenice 5/A35/2S049,
62500 Brno, Czech Republic
email: tevang.pharm.uoa.gr
tevang3.gmail.com
website: https://sites.google.com/site/thomasevangelidishomepage/
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: wrong_geometry.png)

{kind=link}