Date: Sat, 27 May 2017 23:26:55 +0000
Hello,
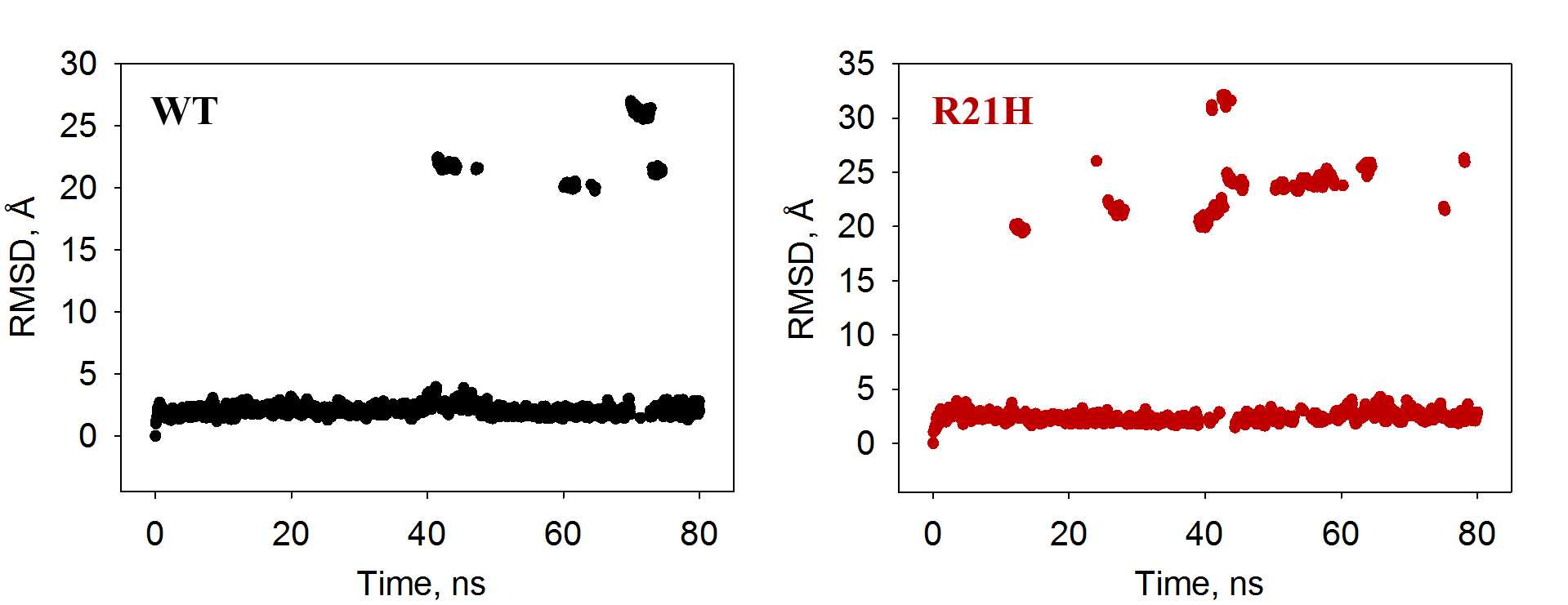
I used Amber 11 to run simulations on two polypeptides. Each polypeptide is a dimer (coiled coil) and the only difference between them is that one has a mutation at residue 21 (R21H) while the other does not (wild-type). No ligand was involved. When I calculated the RMSD, I noticed the "jumps", so I recorded a movie for each simulation and saw that the two strands came apart at certain time points. Attached are scatter plots of RMSD calculations.
My questions are as follows:
1. Is this purely an issue of periodic boundary conditions. If it is, what is the best way to correct it? I'm thinking to re-run each simulation with a bigger solvation box? The one I used was a 10-angstrom water box (the command line was: solvatebox xyz TIP3PBOX 10. 0.75)
2. Despite the jumps, is it still correct to say that the peptides did reach pseudo-steady states? If so, can I just remove the jumps from the RMSD plots without re-running the simulations?
I really appreciate any help and/or suggestions!
Regards,
-- Thu (Lily) Ly Graduate Research Assistant Gene and Linda Voiland School of Chemical Engineering and Bioengineering Washington State University Wegner Hall 334 Pullman WA 99164-6515 Mobile | 206.446.1768 thu.ly.wsu.edu
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: RMSDs.png)
