Date: Sun, 14 May 2017 17:13:54 +0000 (UTC)
Dear All,I have plotted dimer rmsd using following command;parm *.prmtoptrajin md_simulation?.mdcrdtrajin md_simulation??.mdcrdautoimagestrip :Na+,WAT outprefix nowater
rms first out rmsd.dat :1-534.C,CA,Ntrajout AD-chain_complex-reimaged-20ns.nc netcdf
I also tried another command which was suggested in amber archive (http://archive.ambermd.org/201109/0749.html) to improve rmsd;
parm *.prmtoptrajin md_simulation?.mdcrdtrajin md_simulation??.mdcrdcenter :1-267 mass origin
image origin center familiarcenter :1-534 mass originimage origin center familiarstrip :Na+,WAT outprefix nowaterrms first out rmd.dat :1-534.C,CA,Ntrajout AD-chain_complex-reimaged-20ns.nc netcdf
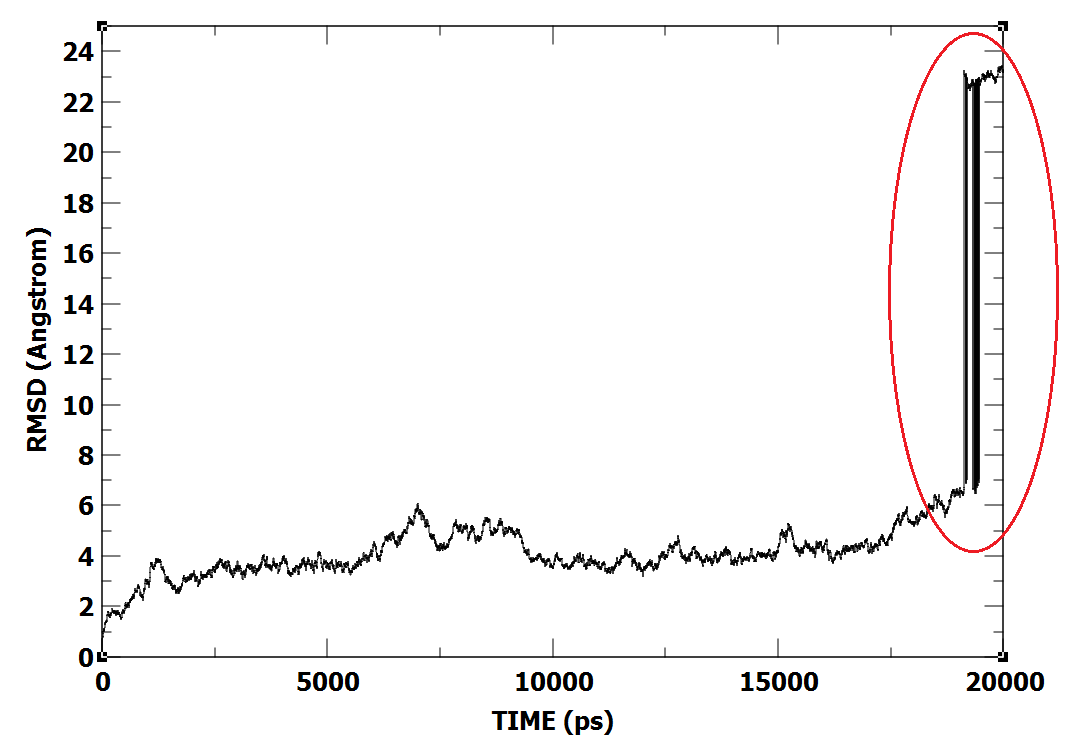
got same result. RMSD plot image attached here. Does anybody know how I might fix the RMSD values Any suggestion would be really appreciated.
Thanks
|
|
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: dimer-rmsd.png)
