Date: Tue, 28 Mar 2017 12:49:36 +0000
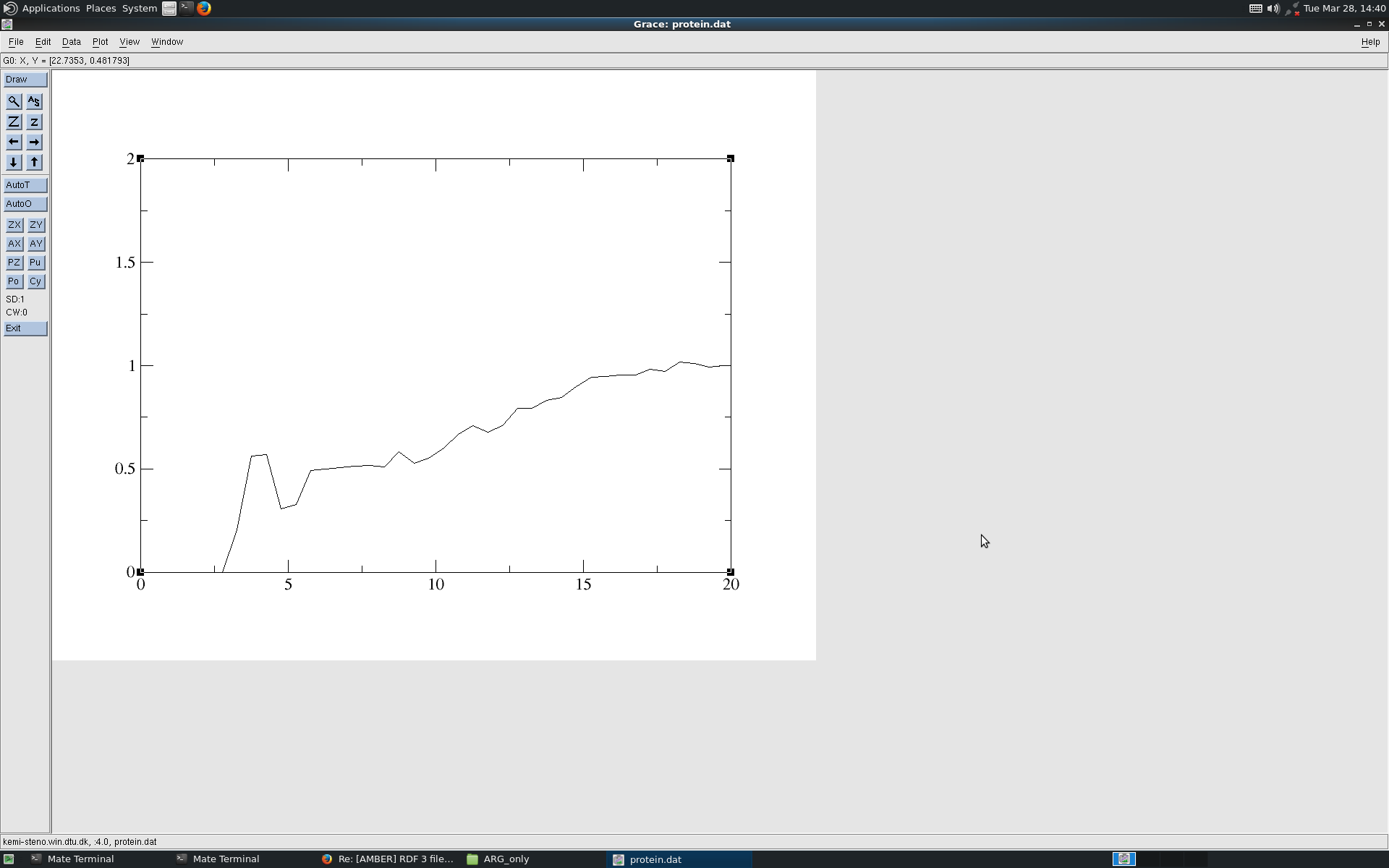
Hi, Thanks for your comments. I did try with one specific atom of a residue (see attached file), it does flatten to 1 but at larger distances. When I reduce the mask :(1-4.CA), it still shows the integrated graph as earlier. Is there way to consider the entire peptide? Thanks. REgards Sowmya ________________________________________ From: Gustavo Seabra [gustavo.seabra.gmail.com] Sent: Tuesday, March 28, 2017 1:26 PM To: AMBER Mailing List Subject: Re: [AMBER] RDF plot Hi, Have you tried to specify just one residue in the protein? Your mask (:1-40.CA,C,N,O) is very large -40 residues-, and I’d expect to see a diffuse pdf, as you show. I believe cpptraj is centering the rdf in the center of mass of the mask, and then counting from there. However, this center is too buried in the protein. — Gustavo Seabra. > Em 28 de mar de 2017, à(s) 07:35, Sowmya Indrakumar <soemya.kemi.dtu.dk> escreveu: > > Dear All, > I am trying to calculate water/solute-peptide radial distribution function g(r). I was successful in calculating the same for the water-water system and water-Cl-. (see the attached file rdf.png). > > 1) radial *.rdf 0.5 15 :WAT.O :WAT.O volume > 2) radial *.rdf 0.5 15 :WAT.O :Cl- volume > > But I am unable to calculate w.r.t to protein or any specific residue in the protein. (see attached file: protein) > I used the below command: > > 3) radial *.rdf 0.5 15 :WAT.O :1-40.CA,C,N,O volume > > Kindly, let me know. > Regards > Sowmya > > <rdf.png><protein>_______________________________________________ > AMBER mailing list > AMBER.ambermd.org > http://lists.ambermd.org/mailman/listinfo/amber _______________________________________________ AMBER mailing list AMBER.ambermd.org http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: residue_2.png)
