Date: Fri, 17 Mar 2017 21:09:32 +0800 (CST)
Dears:
Thanks for your replay!
I have a question: some dummy VdW parameters for QM region will give a reliable QM/MM MD results?
If use dummy VdW parameters, the second term in the [ ] will be incorrect.
Best wishes!
At 2017-03-17 03:08:50, "David Poole" <thepoole.ucdavis.edu> wrote:
>Hello,
>
>I'd note that the older version of parmchk2 (AmberTools15) and the current
>release of parmchk will put dummy parameters for non-library atoms.
>
>After adding atomic masses and some dummy VdW parameters they can be used
>for QMMD and QM/MM/MD with amber if they are included in the QM region.
>
>It might be nice if there was an option in parmchk2 to suppress/ignore
>errors like this just to have a properly formatted frcmod file.
>
>-DP
>_______________________________________________
>AMBER mailing list
>AMBER.ambermd.org
>http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
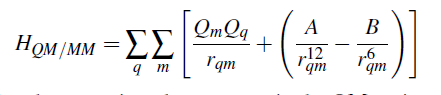
(image/png attachment: aa.png)
