Date: Wed, 30 Nov 2016 14:58:45 +0100
Dear Amber Users,
I found an issue with the gaff parameters for "nh" atom type assigned to
the amino nitrogen, connected to aromatic ring.
I am doing MD simulation of NADP molecule in TIP3P water box at NPT
ensemble.
The NADP molecule is parameterized using GAFF force field.
I got charges for NADP from RED server.
In antechamber, the amino nitrogen connected to aromatic ring is assigned
as "nh" atom types.
antechamber -i NADP_RED.mol2 -fi mol2 -o NADP_antechamber.mol2 -fo mol2
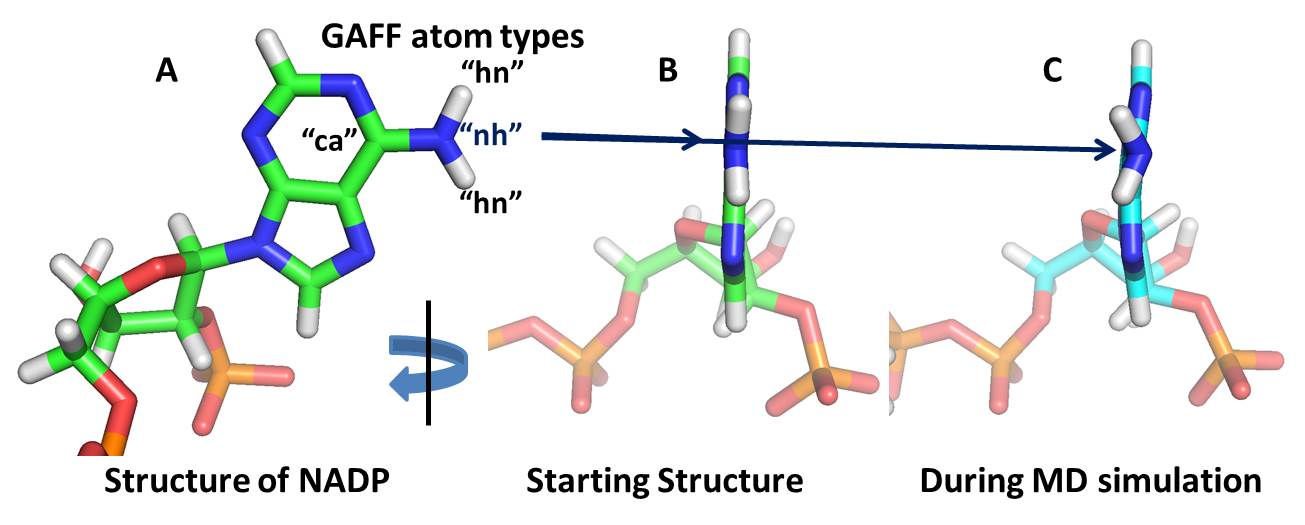
Initially (before simulation), the amino group has planner geomerty and is
in the same plane of aromatic ring. (As shown in attached image A and B).
But during MD simulation, this amino group has distorted geomerty and
oscillating out of the plane. (As shown in attached image C).
As far i understand, this Nitrogen atom should have sp2 hybridized state,
and amino group must be in the same plane of aromatic ring with trigonal
planar geometry.
I observed the same phenomenon for other ligands with
similar amino group (gaff atom type "nh") connected to aromatic ring.
But such distortion was not present in amide
group of NADP, where Nitrogen has gaff atom type "n" (for sp2 nitrogen in
amide): here it has planner geometry.
1) Is it normal behaviour of amino group attached to aromatic ring?
2) I don't understand Why it is behaving like sp3 nitrogen or i
misunderstood it?
So kindly, I request you to please clarify the doubt, if you come accross
similar observation.
Thanks and Regards,
---------------------------------------------
Dr. Prajwal P. Nandekar
Postdoc, Molecular and Cellular Modeling
HITS, Heidelberg
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: nadp_gaff_issue.jpg)
