Date: Thu, 2 Jun 2016 08:25:53 +0800 (CST)
Hello,
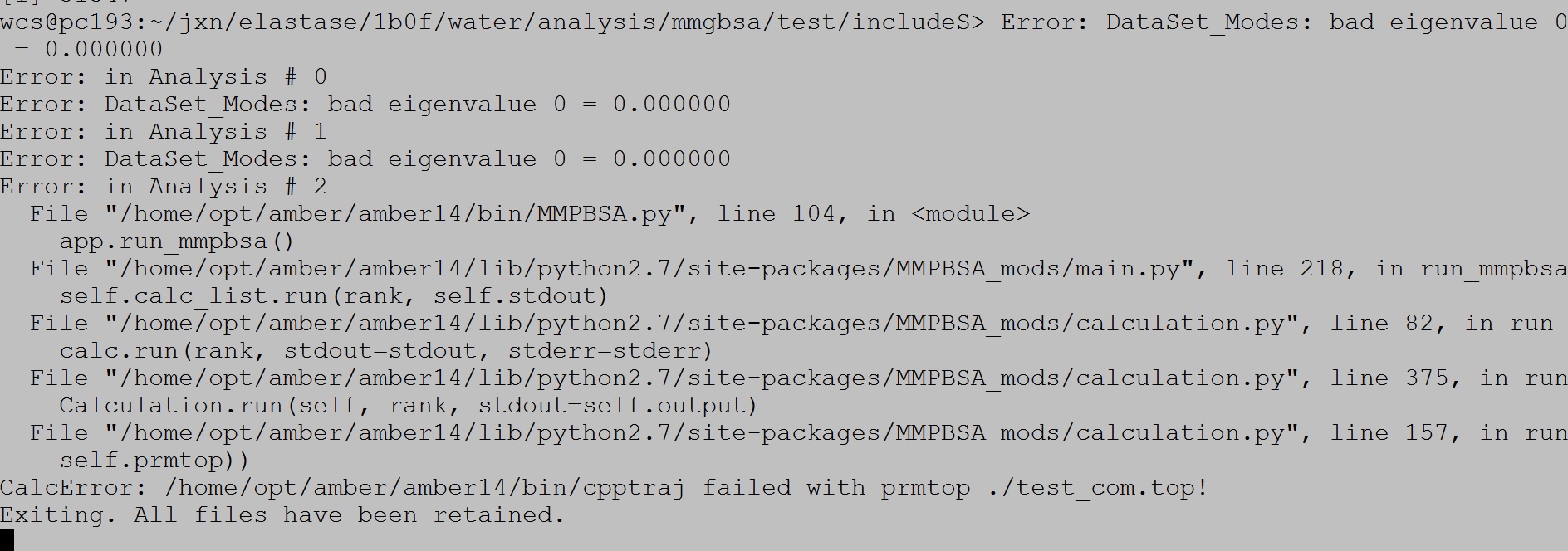
I want to calculate the binding energy of a complex using Ambertools. Firstly, I did not take the entropy into consideration. I got the result file. Then, I put the "entropy=1" keyword into the input files. Some errors appeared. How can I deal with this problem. Help me please. I upload my input files and the error picture. Thank you very much!!!
Best Wishes!
Canaan
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: error.jpg)
- application/x-sh attachment: gbsa.sh
- application/octet-stream attachment: mmpbsa.in
