Date: Thu, 26 May 2016 19:18:56 -0500
Hi Dear Amber users, I am calculating binding free energy for a peptide (25
aminoacids) and a ligand (Creatinin, which I got parameters from
RED-tools).
I use ubuntu 14.04, and pmemd.cuda with a GTX 970.
I follow procedure described in tutorial 3, I changed parameters for
"restrained_mask" to '26', according to my molecules, but I kept the same
parameters as in tutorial. when I convert to pdb in order to see changes in
my molecules, I noticed a change when made equilibration (equil.in), this
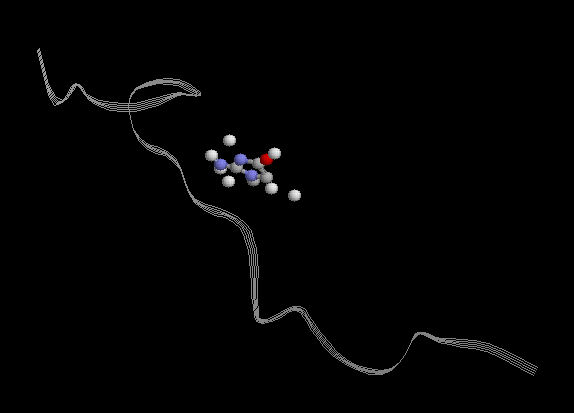
is because in equil.in input file there is no restraints, but my ligand is
totally broken as shown in figure attached, and pdb files form productions
are equal, ligand is far away from its original site and separated.
My question is, are parameters in tutorial 3 convenient for my calcules,
using a molecule like creatinin?
I give thanks in advance
Regards
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: ligando_en_posicion_original.png)

(image/png attachment: ligando_disperso.png)
