Date: Wed, 4 May 2016 10:28:34 +0200
Hello Conor,
I got curious about your molecule and took a look at it. I did some quick
HF/6-31G(d) calculations and found two minima that were close in energy -
one that is the conformation you sent out, and a second lower energy (by
0.43 kcal/mol) conformation where the amide group functional group is
rotated 180 degrees (see attached figure that contains the AM1-BCC charges
for this conformation). I verified that they are minima on the potential
energy (PE) surface by doing frequency calculations. I also did two sets
of torsion rotations, one for each functional group of the phenyl ring. I
then compared how the gaff parameters perform using the AM1-BCC charge set
(see attached PE figures). The rotation of the hydroxyl group looks decent,
in my opinion. However, the rotation of the amide functional group deviates
significantly from the HF/6-31G(d) results. Around both HF/6-31G(d) minima
(0 and 180 degrees), the molecular mechanics (MM) calculations predict a
double well potentials.
I would say these are preliminary results. To really verify the QM
surface is reliable, I would suggest doing the calculations at a more
rigorous theory level (e.g. MP2/aug-cc-pVDZ or MP2/cc-pVTZ). Right now, I
don't truly know if the HF/6-31G(d) or MM results are correctly describing
the shape of the PE curves. (However, from experience I believe that the QM
results are correct.)
The question now becomes is how important are these small conformational
changes for your investigation. If they are very important, then you might
have to do some parameter optimization.
Hope this helps in thinking about your model.
Bests,
Karl
On Tue, May 3, 2016 at 10:13 PM, conor parks <coparks2012.gmail.com> wrote:
> David,
>
> thank you very much for providing that link. That was very helpful. That
> clears up the negative sign issue for me. Thanks! Was my first question,
> correct? I.E. the dihedral potential and forces get evaluated for each
> parameter set associated with it, even when there are multiple, and the
> results are summed?
>
> Regards,
>
> Conor Parks
> B.S.E in Chemical Engineering, University of Michigan, 2012
> PhD candidate in Chemical Engineering, Purdue University
>
> On Tue, May 3, 2016 at 3:53 PM, David A Case <david.case.rutgers.edu>
> wrote:
>
> > On Tue, May 03, 2016, conor parks wrote:
> > >
> > > However, in my prmtop file (values given in first email in this chain),
> > the
> > > periodicity values are given as 1 and 2 (no negative sign). What
> happened
> > > to the negative sign on these periodicity values in my prmtop file?
> >
> > Please see http://ambermd.org/formats.html#parm.dat. Negative values
> in a
> > parm.dat file have a special meaning.
> >
> > ...dac
> >
> >
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- Karl. N. Kirschner, Ph.D. Research Associate Bonn-Rhein-Sieg University of Applied Sciences Grantham-Allee 20, 54757 Sankt Augustin, Germany
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
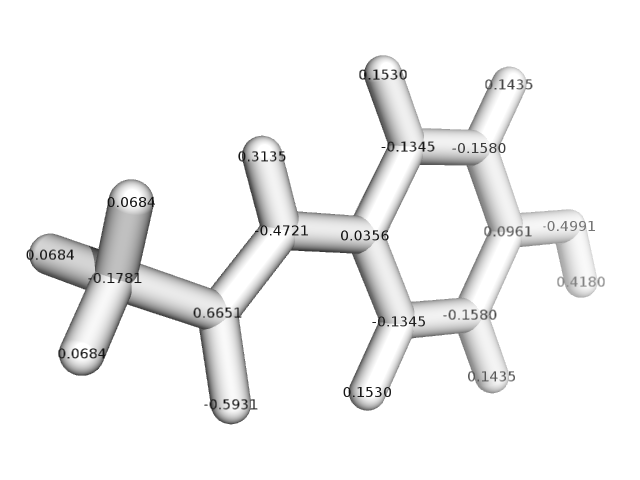
(image/png attachment: bcc_charges.png)
- application/pdf attachment: Figure_E_ca-ca-oh-ho.pdf
- application/pdf attachment: Figure_E_ca-ca-n-c.pdf
