Date: Tue, 12 Apr 2016 10:37:55 +0530
Can anyone explain what this 2.0 :2 denotes in this command
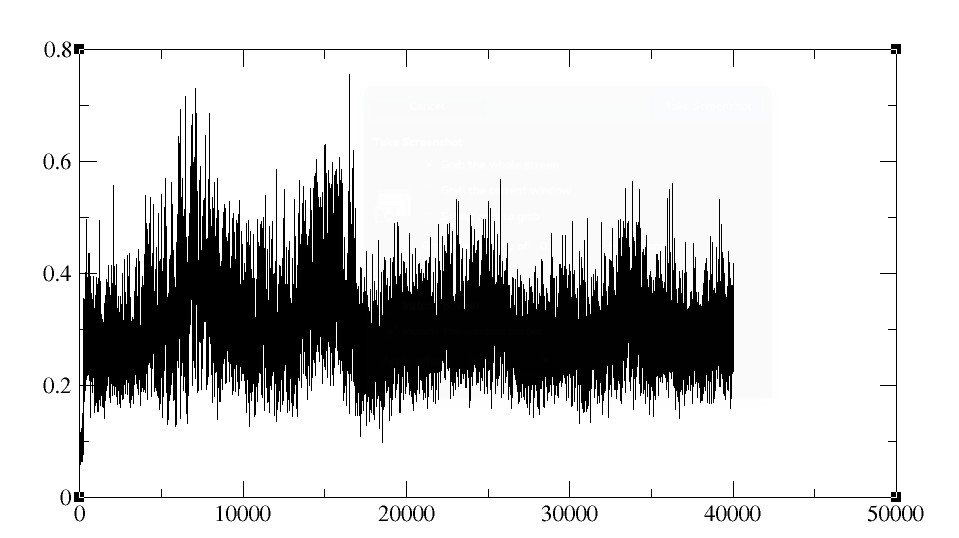
"rms reference mass out 02_03.rms time 2.0 :2"
I have used time step of 2 fs to run the simulation and ran upto 4ns and
tried to plot the rmsd graph (mass weighted) using the command below
"rms first mass out mass.rms time 2.0 :2"
and got the graph like this. Can anyone address what's going wrong in this
On Fri, Apr 8, 2016 at 9:44 PM, Daniel Roe <daniel.r.roe.gmail.com> wrote:
> If you just want the backbone RMSD you will need to modify your atom
> masks to only include backbone atoms; ':1-199' selects all atoms from
> residues 1 to 199. For your protein residues, you could do something
> like ':1-177.C,CA,N' etc. See the Amber 15 manual section 29.1.6 for
> more information and examples of the Amber atom mask selection syntax.
>
> -Dan
>
> On Fri, Apr 8, 2016 at 5:04 AM, Sreemol G <sreemolinfo.gmail.com> wrote:
> > I need to calculate backbone RMSD for Protein-RNA complex. the command
> > given below is the one i used to calculate backbone RMSD, I would like to
> > know whether it is sufficient to cover all the information about both
> > protein and RNA backbone. I have 199 residues in my complex. Among them
> 177
> > are amino acids and 22 are RNA.
> >
> > trajin md1.mdcrd
> > trajin md2.mdcrd
> > rms first out backbone.rms :1-199
> >
> >
> > On Fri, Apr 8, 2016 at 3:00 PM, Dr. Anselm Horn <anselm.horn.fau.de>
> wrote:
> >
> >> Dear Sreemol,
> >>
> >> > I have done simulation for protein-RNA complex for 10 ns, I want to
> >> > calculate the backbone RMSD for the complex. How to calculate the
> >> backbone
> >> > RMSD for protein-RNA complex.
> >>
> >> backbone RMSD calculations are a very basic analysis technique. For such
> >> analyses, many people have created excellent tutorials available from
> >> the amber webpage ambermd.org. A first example of the RMSD analysis is
> >> given in the very first tutorial:
> >>
> >> http://ambermd.org/tutorials/basic/tutorial0/
> >>
> >> The second tutorial deals with DNA, so you might also want to have a
> >> look at that.
> >>
> >> Additionally, the documentation of the Amber suite contains a detailed
> >> description of all programs and input parameters (cf
> >> http://ambermd.org/doc12/).
> >>
> >> All the best,
> >>
> >> Anselm
> >>
> >> Dr.rer.nat.
> >> Bioinformatik
> >> Friedrich-Alexander-Universität Erlangen-Nürnberg
> >> Germany
> >>
> >>
> >> _______________________________________________
> >> AMBER mailing list
> >> AMBER.ambermd.org
> >> http://lists.ambermd.org/mailman/listinfo/amber
> >>
> >
> >
> >
> > --
> > With kind regards,
> > G. Sreemol
> > M.Tech (Computational Biology)
> > Anna university
> > Chennai.
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
>
>
>
> --
> -------------------------
> Daniel R. Roe, PhD
> Department of Medicinal Chemistry
> University of Utah
> 30 South 2000 East, Room 307
> Salt Lake City, UT 84112-5820
> http://home.chpc.utah.edu/~cheatham/
> (801) 587-9652
> (801) 585-6208 (Fax)
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- With kind regards, G. Sreemol M.Tech (Computational Biology) Anna university Chennai.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: Screenshot_from_2016-04-12_10-25-00.jpg)
