Date: Sun, 10 Apr 2016 15:17:32 -0300 (BRT)
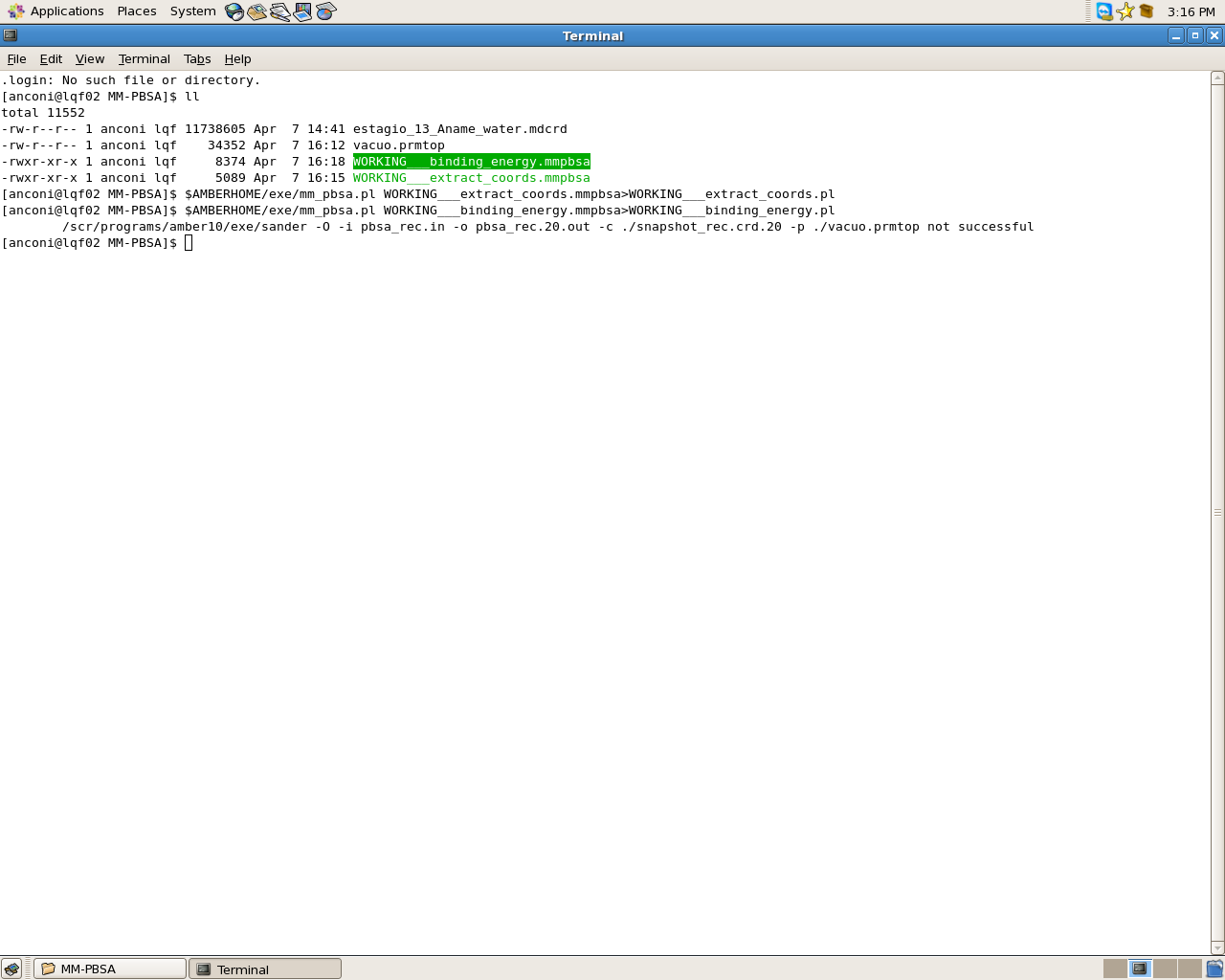
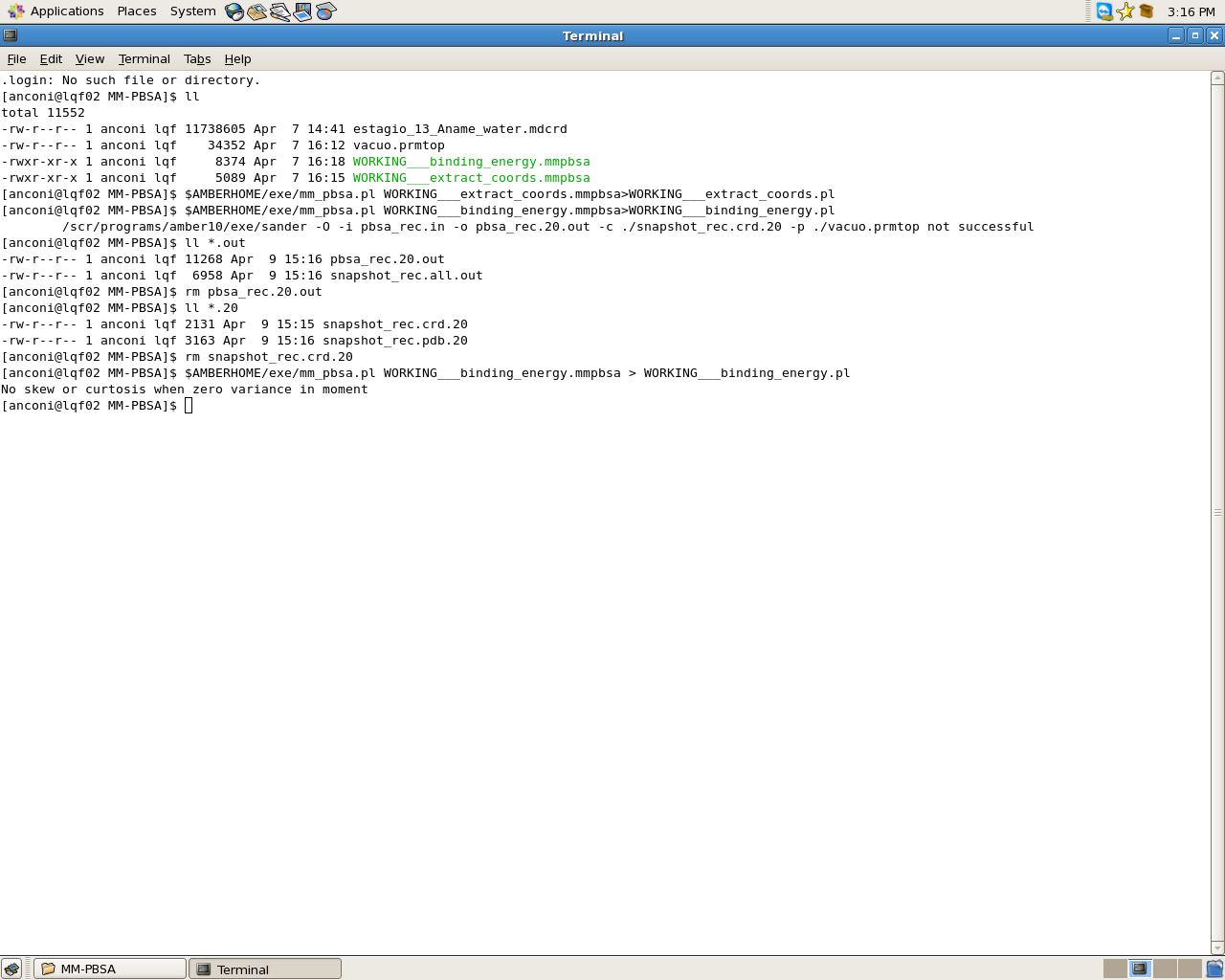
I'm having this problem that I have attached the picture, when I delete the file pbsa_rec.20.out and snapshot_rec.crd.20 and use the command again = $ AMBERHOME / exe / mm_pbsa.pl WORKING ___ binding_energy.mmpbsa > WORKING___binding_energy.pl I get results , that I 'm doing this right?
Attached files.
The molecule is a tetracycline and the system is the tetracycline complexation with B-cyclodextrin for B-cyclodextrin and the complex I can usually get the results but for tetracycline happens mentioned above.
I appreciate the attention.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- application/octet-stream attachment: WORKING___extract_coords.mmpbsa
(image/png attachment: Screenshot-1.png)
(image/png attachment: Screenshot-2.png)
- application/octet-stream attachment: snapshot_statistics.out
- text/x-matlab attachment: vacuo.prmtop
- application/octet-stream attachment: WORKING___binding_energy.mmpbsa
