Date: Fri, 1 Apr 2016 08:03:06 +0000
Dear Amber experts,
I have Amber14 and Amber Tools 15 on my linux computer.
Following protocols from corresponding Amber tutorials I have simulated
membrane protein by conventional MD (cMD) and accelerated MD (aMD).
Regarding aMD I had a question: Do I need to account for setting EthreshD
and alphaD parameters in the production MD script the residues of
protein only or both - protein and membrane?
There are alternatives:
Protein only.
In this case the job was immediately stopped with the message in nohup.out file:
cudaMemcpy GpuBuffer::Download failed an illegal memory access was encountered
Accounting for both - protein and lipid residues the aMD production job runs smoothly.
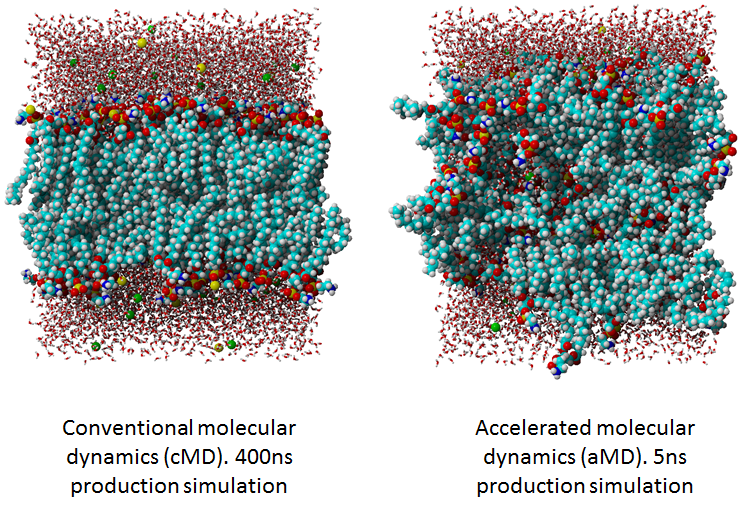
But there is one small problem: aMD completely ruins membrane structure already after
couple of ns simulation time in contrast to conventional MD.
See for illustration figure below presenting the membrane structure after
400ns production run of cMD vs. 5ns of aMD.
Don't you think that there should be an option in amber software allowing to account for
protein residues only, but not membrane in the setting of EthreshD and alphaD parameters
for the aMD production phase?
Thank you,
Michael
[cid:cda95163-983d-45c8-8a45-4431784c1029]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: pastedImage.png)
